Полное секвенирование экзома в г. Москва
Полное секвенирование экзома — это комплексное генетическое тестирование, в котором используется технология секвенирования нового поколения (NGS) для анализа кодирующих областей генома
20 000 генов. Данные кодирующие области называются «экзонами», а все экзоны вместе называются «экзомом». Поскольку при полном секвенировании экзома исследуется большое количество генов, данный метод позволяет обнаружить причину заболевания в тех случаях, когда другие исследования не дали результата.
Зачем делать полное секвенирование экзома
Данный вид исследования помогает установить диагноз и спрогнозировать дальнейшее течение болезни и определить риски возникновения осложнений или их отсутствие.
Правильное определение мутаций и понимание диагноза дает возможность пациенту совместно с врачом генетиком выработать стратегию действий направленную на предупреждение осложнений. По результатам исследования врач может определить риски возникновения генетических заболеваний у будущих детей.
А также совместно с пациентом скорректировать планирование беременности с помощью ЭКО и подобрать ряд диагностических исследований: ПГТ-М, НИПТ и тд
Точность генетического анализа
Полное секвенирование экзома позволяет выявить определенные мутации в генах, которые были унаследованы от родителей либо появились спонтанно.

Анализ проводится по венозной крови пациента. С помощью метода секвенирования NGS на полногеномном секвенаторе MGISEQ-2000 (BGI inc.) точность прочтения каждого фрагмента ДНК для >85% прочтений достигает 99,98%. Средняя глубина покрытия ≥100 x, это значит, что каждая буква в геноме была прочитана минимум 100 раз.
Показания для полного секвенирования экзома
Как тест первой линии, это исследование может быть назначено:
Можно ли сдать этот анализ здоровому человеку?
Да, можно. Даже если у человека нет никаких признаков заболеваний и осложнений, он может сделать такой анализ ДНК. Результаты исследования дадут пациенту понимание где и в каких генах есть мутации.
Такой скрининг на носительство мутаций подойдет для людей, которые планирую беременность.
Вторичные находки
Во время анализа секвенирования могут быть обнаружены некоторые варианты, которые не связаны с текущими симптомами пациента (вторичные находки). Тем не менее, эта информация может иметь важные последствия для здоровья пациента и членов их семей.
Вторичные находки могут быть включены в отчет пациента. Наше секвенирование экзома включает все гены, перечисленные в списке генов ACMG (Американский колледж медицинской генетики и геномики). Мы учитываем последние рекомендации по генетическому тестированию, чтобы улучшить качество наших заключений. Список генов ACMG (скачать)
Как сдать анализ на полное секвенирование экзома
После получения заключения обязательно покажите его врачу-генетику. Мы не экономим на здоровье наших пациентов! Поэтому срок исполнения анализа 45 дней. Мы не увеличиваем срок анализа, чтобы “накопить образцы” и удешевить анализ. Поэтому сроки в лаборатории Медикал Геномикс строго регламентированы.
В команде Медикал Геномикс работают врачи-генетики, имеющие ученые степени в биологии и медицине, которые постоянно повышают квалификацию, публикуют научные статьи в цитируемых журналах и являются членами РАРЧ, РОМГ, ESHRE.
Команда биоинформатиков анализирует данные секвенирования экзома с 2010 года и имеет огромный опыт в биоинформатической обработке данных.
Результат теста ДНК полного секвенирование экзома
Данный анализ выявляет:
Полное секвенирование генома что это
Сегодня много пациентов страдает от неверного диагноза или несвоевременно обнаруженного заболевания, связанного с нарушениями в структуре ДНК.
Распространенные генетические тесты, такие как анализ одного гена, панели из нескольких генов или микроматричный анализ, часто не могут до конца выявить точную причину болезни из-за своих ограниченных возможностей.
Недавние достижения молекулярной генетики позволили сделать доступным по цене и скорости выполнения новый метод генетического тестирования — полногеномное секвенирование.
Полное секвенирование генома способно обнаружить почти все изменения в ДНК пациента, расшифровывая последовательность всех кодирующих и некодирующих областей. Результат такого исследования — информация о тысячах генов, участвующих в нормальном росте и развитии организма.
Этот метод с успехом заменяет все ранее известные способы генетического тестирования, соединяя в себе их возможности. С каждым годом он позволяет установить точный диагноз все большего числа заболеваний.
Используя полногеномное секвенирование, лечащий врач получает помощь не только в диагностике, но и более точно принимает решения по лечению, может наблюдать за течением заболевания, делать прогноз развития болезни и выздоровления. Точные рекомендации о прогрессировании заболевания также возможны в некоторых случаях.
По результатам тестирования может быть проведена оценка риска наследственных заболеваний для других членов семьи.
Полное секвенирование может быть проведено по направлению лечащего врача, а также по желанию пациента без наличия срочных медицинских показаний.
Структура генома не изменяется в течение всей жизни. Сделав анализ однократно, его результатами можно пользоваться всю жизнь. К ним можно повторно обращаться при появлении очередных открытий в генетике и с учетом новых знаний более точно интерпретировать данные.
Имея на руках готовую расшифровку своего генотипа, человек подготовит себя к возможным экстренным ситуациям, когда подробная генетическая информация может спасти жизнь — травмы, операции, тяжелые заболевания, пересадка органов и другие.
Полногеномное секвенирование — лучший на сегодня метод продолжать исследование структуры ДНК, функций генов, их влияния друг на друга и на проявления фенотипа.
Хотя крупные исследовательские центры в мире ведут эту работу, ее результаты не всегда широко доступны.
Поэтому многие исследователи делают это самостоятельно для своих узкоспециализированных целей — подбор и разработка препаратов, спортивные достижения, селекция растений и животных, диагностика и лечение болезней.
Лаборатория «Геномед», обладая современным оборудованием, проверенными технологиями и квалифицированными специалистами-генетиками, готова предоставлять эти ресурсы для целого ряда научных разработок.
Фундаментальные исследования
Изучение структуры и функции генома
Исследование генома человека не завершено до сих пор. Полногеномное сенквенирование дает новые возможности поиска нераскрытых функций генов, изучения некодирующих областей и регуляторных участков ДНК.
Структурные изменения и CNV
В отличие от большинства других генетических методов полногеномное секвенирование позволяет точно определять любые изменения структуры ДНК.
Часто достигается точность определения до уровня единичного нуклеотида. Поэтому, если требуется достоверно определить транслокации, инверсии, вставки или делеции участков, стоит использовать тест Genome UNI.
Эпигенетика
После создания технологии полногеномного секвенирования появилась возможность изучать закономерности наследования, не связанные с генетической последовательностью. В первую очередь речь идет о механизмах регуляции экспрессии генов.
В вопросах эпигенетики важную роль играют методики исследования метилирования ДНК (MeDIP-Seq, WGBS), взаимодействия и модификации гистонов (ChlP-Seq), метилирования РНК (TRM).
Полногеномные ассоциативные исследования
Исследование связи между структурой генома и фенотипическими признаками позволяет выявить, как проявляются различные сочетания генов.
Результат такой работы — более четкое понимание механизмов развития полигенных и многофакторных заболеваний, выявление влияния гетерозиготности на развитие признаков и оценка влияния средовых факторов.
Митохондриальный геном
В отличие от других генетических тестов полногеномное секвенирование исследует одновременно весь генетический материал клетки — ДНК ядра и ДНК митохондрий. Это особенно важно при изучении наследственности по материнской линии и обнаружении наследственных митохондриальных заболеваний.
Транскриптом
Полногеномное секвенирование позволяет оценивать не только геном клетки, но и ее транскриптом — совокупность всех типов РНК.
Важность транскриптома в том, что он показывает активность экспрессии определенных генов на текущий момент. Эта активность сильно зависит от состояния окружающей среды и самого организма и может говорить о наличии неблагоприятных процессов.
Биоинформатический анализ
Расширенный пайплайн
Многопотоковое секвенирование позволяет одновременно проводить расшифровку множества участков одной ДНК. Это значительно ускоряет процесс чтения, позволяет снизить стоимость и добиться высокой точности.
В настоящее время полногеномный анализ покрывает больше 96% генов с глубиной прочтения >20х, а время исследования одного генома сократилось до 1 суток.
Платформа управления данными
Результаты анализов представлены в виде удобного пользовательского интерфейса в личном кабинете. Там же доступны данные интерпретации, если она проводилась, и полученные рекомендации.
Владелец доступа может в любое время обратиться к своим результатам или загрузить их для off-line использования.
Пользовательский биоинформационный сервис
Кроме оборудования и передовых технологий «Геномед» рад предоставить консультационные услуги опытных врачей-генетиков.
Направляя пациента на анализ, вы получите не только расшифровку нуклеотидной последовательности, но и, при необходимости, ее клиническую интерпретацию и рекомендации по использованию результатов.
Собственное хранилище данных
Данные о проведенных исследованиях хранятся на собственных серверах лаборатории.
Все данные шифруются и доступ к ним предоставляется исключительно заказчику с индивидуальным логином и паролем.
Прикладные исследования
Диагностика генетических заболеваний
Сегодня насчитывают около 6000 различных генетических заболеваний и нарушений. Многие из них обладают схожими симптомами, но вызваны патологией разных генов. Если искать эти заболевания по отдельности, потребуется огромное количество исследований.
Полногеномное секвенирование позволяет заменить все виды генетических и многих лабораторных исследований единственным точным и высокоинформативным тестом.
Геном опухоли
Полногеномное секвенирование опухоли дает ключ к пониманию канцерогенеза. Это не только определение основных мутаций, приводящих к развитию рака.
Получив полную расшифровку генома, онколог может оценить уровень сопротивляемости организма росту опухоли, следить за изменением опухоли под воздействием терапии, подбирать наиболее эффективные и безопасные сочетания препаратов.
Профилактическое здоровье
Данные об индивидуальных предрасположенностях к заболеваниям могут изменить образ жизни человека.
На этом основании проще подобрать диету, нагрузки, препятствовать или способствовать воздействию различных факторов внешней среды.
Это позволит не просто увеличить продолжительность жизни, но и улучшить ее качество.
Создание лекарств
Данные об индивидуальных предрасположенностях к заболеваниям могут изменить образ жизни человека.
В зависимости от своего генетического статуса человек по разному реагирует на лекарственную терапию. Гены влияют как на эффективность, так и на переносимость препаратов. Полногеномное тестирование позволяет решать две важные задачи фармакотерапии.
Первая задача — поиск биомаркеров заболеваний. Наглядный пример — простатоспецифический антиген, используемый для диагностики опухолей предстательной железы.
Вторая задача — персонализация лекарственной терапии. В идеале препарат должен разрабатываться для генотипически близких групп пациентов или даже индивидуально с учетом конкретного набора генов.
Судебная медицина / Судебная патология
Генетические данные сегодня широко используются в криминалистической и судебной практике. Ниже перечислены некоторые примеры такого применения. Определение родства\материнства\отцовства.
Антропология и этнос
Изучение ДНК предков или представителей различных этнических групп позволяет понимать историю происхождения и развития человека и законы передачи наследственной информации.
Для отдельного человека появляется возможность разобраться в своих корнях и родословной. Для общества и науки — это источник изучения этнических особенностей здоровья, заболеваемости, долголетия и возможностей развития.

Диагностика наследственных заболеваний
Анализ всего генома в одном исследовании чаще всего приводит к более быстрой постановке диагноза генетического состояния по сравнению с многочисленными сериями одиночных генетических тестов. Особенно это справедливо, если клиническая картина не дает генетику четких указаний, в каких локализациях следует искать патологию, либо тогда, когда клиника может быть обусловлена патологией разных или нескольких генов.
Репродуктивное здоровье
Четко определена связь ряда генов с репродуктивной функцией. Мужское и женское бесплодие, невынашивание, хромосомные болезни новорожденных — все эти состояния можно обнаружить, а впоследствии и успешно лечить, если знать состояние генов, ответственных за процессы зачатия и развития плода.
Онкология
Молекулярно-генетические исследования — мощный инструмент в борьбе с онкологическими заболеваниями.
Во-первых, расшифровав полный геном, можно оценить не только гены, напрямую связанные с развитием рака. Одновременно врач получает полную картину состояния антиоксидантной защиты, противоопухолевого иммунитета, антитоксической функции и других систем, которые косвенно влияют на появление и течение онкопроцесса.
Во-вторых, генетические методы позволяют более эффективно подобрать препарат, контролировать процесс лечения и склонность к рецидивированию.
Важно, что полногеномное исследование (WGS) экономически намного выгоднее, чем исследование отдельных генов или серии генетических панелей (наборов анализов генов, связанных с определенной патологией).
Сегодня с помощью полногеномного тестирования можно оценить 649 опухолевых генов и еще 28 генов, способных нести хромосомные транслокации (перенос участков ДНК между хромосомами), связанные с развитием рака.
Планирование семьи
Известно, что предрасположенность к заболеваниям передается по наследству. Существует группа заболеваний, которые зависят от состояния генов и носят название аутосомно-рецессивных.
Полногеномное секвенирование способно показать людям, вступающим в брак, риски рождения у них детей с аутосомно-рецессивными болезнями.
Предрасположенность к заболеваниям
Почему одни люди заболевают раком, а другие нет? Кто-то живет до 100 лет, многие же умирают в молодости. Как узнать причину?
Сегодня уже четко установлена связь многих генов с развитием заболеваний. Причем, состояние одних генов улучшает прогноз, других — ухудшает. Примеры таких болезней — атеросклероз, остеопороз, тромбозы.
Если знать сочетание всех генов, можно рассчитывать риск развития заболеваний для человека, даже если он пока абсолютно здоров, а также предотвращать их появление.
Индивидуальные способности
Для многих способностей и склонностей человека показана прямая зависимость от состояния его генотипа. К ним относятся мышечная выносливость и сила, темперамент, наклонность к разного рода зависимостям и многое другое. Если планировать профессию и образ жизни на основании генетического анализа, можно достигать лучших результатов быстрее и с меньшими усилиями.
Почему нужно использовать
секвенирование генома
Полногеномное тестирование обладает целым рядом преимуществперед другими методами
в скорости, широте и точности расшифровки генетической последовательности
и установлении диагноза.
| Секвенирование панелей генов | Клиническое секвенирование экзома | Полное секвенирование экзома | Genome UNI | |
|---|---|---|---|---|
| Покрытие кодирующих участков генома | Равномерное, только в области экзонов исследуемых генов | Неравномерное | Неравномерное | Равномерное по всему геному |
| Поиск вариантов в интронах | — | — | — | + |
| Поиск вариантов мтДНК | — | — | — | + |
| Определение CNVs с высокой точностью | — | — | — | + |
| Определение экспансии тринуклеотидных повторов | — | — | — | + |
| Вероятность выявления причины заболевания при повторном анализе данных | Низкая | Низкая | Средняя | Высокая |
| Показания к исследованию | Заболевания с преимущественным поражением одной системы органов или одним ведущим симптомом, когда определены гены ассоциированные с фенотипом | Подозрение на определенную генетическую патологию, когда другие методы (анализ отдельного гена или панели) недоступны | Секвенирование пробанда или родителей (трио) для поиска генов кандидатов (GUS) при отсутствии патогенных вариантов в клинически значимых генах | «В качестве теста первой линии у пациентов с признаками наследственного заболевания или при отсутствии специфического фенотипа.» |
| Другие особенности | Повышенное покрытие таргетных участков генома позволяет лучше выявить однонуклиотидные и in/del варианты | Нет рекомендованного перечня клинически значимых генов | Не обладает преимуществами при секвенировании только пробанда | Большой объем данных требует использования дополнительных вычислительных ресурсов |
Информация для исследователей
Фундаментальные исследования
Секвенирование и ресеквенирование генома, анализ метагенома, экспрессии и метилирования генов.
Прикладные исследования
Поиск биомаркеров, рекрутинг пациентов, решения для селекции.
Медицинские исследования
Секвенирование генома и экзома, анализ на микрочипах.
Биоинформатический анализ
Расширенный пайплайн, биоинформационный сервис, платформа управления данными, хранилище данных.
Genome UNI рекомендован:
Врачам на заметку!
Genome UNI выявит:
Обладает ли тест Genome UNI
доказанной эффективностью?
Для анализа данных используется проприетарный алгоритм и пайплайн, разработанный одной из ведущих компаний США, которая специализируется на анализе данных полного секвенирования генома.
Пайплайн разработан в соответствии с требованиями профессиональной ассоциации медицинских генетиков (ACMG) и ассоциации клинических патологов (CAP) США и включает возможность анализа данных с учетом формализованного описания фенотипа в терминах HPO.
Строгий подход к оценке качества лабораторных данных при использовании профессионального пайплайна позволяет не только повысить выявляемость вариантов, но и избежать ошибок которые могут возникнуть на стадии сиквенса и снизить вероятность ложноположительных результатов.
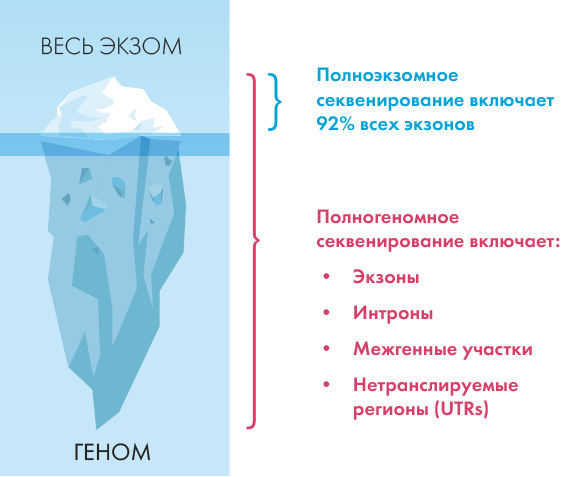
WGS выявляет больше инсерцционно-
делеционных вариантов при 30х чем WES
при средней глубине 100х
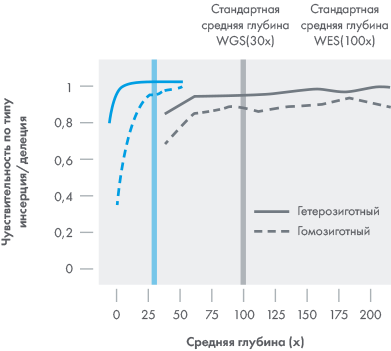
WGS охватывает больше генов на при 30х,
чем WES при 100х
Секвенирование геномов для «чайников»
 Геномика: постановка задачи и методы секвенирования
Геномика: постановка задачи и методы секвенирования
 Геномика: постановка задачи и методы секвенирования
Геномика: постановка задачи и методы секвенированияСергей Николенко, кандидат физико-математических наук, старший научный сотрудник лаборатории вычислительной биологии Санкт-Петербургского Академического Университета в серии статей говорит о некоторых задачах биоинформатики, связанных со сборкой и анализом геномов, делая акцент на математической, комбинаторной постановке задачи. В данном, вводном, тексте речь идет о том, как выглядят входные данные для сборки геномов и как их получают.
Как выглядит молекула ДНК?
Начнем с того, как выглядит молекула ДНК. Молекулы полимеров характеризуются первичной структурой, под которой понимается просто состав молекулы (в данном случае – последовательность букв A, C, G и T, которые и составляют геном), вторичной структурой, т.е. тем, какие именно химические связи устанавливаются между этими компонентами и какие в результате получаются базовые пространственные структуры (в данном случае – двойная спираль), и третичной структурой, т.е. тем, как вторичная структура «уложена» в пространстве. Вторичная структура ДНК представляет собой двойную спираль, состоящую из четырёх разных нуклеотидов.

Рисунок из Википедии
Нуклеотиды обозначаются по содержащимся в них азотистым основаниям: аденину (A), цитозину (C), гуанину (G) и тимину (T) (есть ещё урацил, который в РНК заменяет тимин), и в дальнейшем мы всегда будем пользоваться этими буквами. В двойной спирали эти нуклеотиды связаны друг с другом водородными связями, и связь устанавливается по принципу комплементарности: если в одной нити ДНК стоит A, то в комплементарной нити будет T, а если в одной нити C, то в другой будет G. Именно это позволяет относительно просто проводить репликацию (копирование) ДНК, например, при делении клетки: для этого достаточно просто разорвать водородные связи, разделив двойную спираль на нити, после чего парная нить для каждого «потомка» автоматически соберётся правильно. Важно понять, что ДНК – это две копии одного и того же «текста» из четырёх «букв»; «буквы» в копиях не идентичны, но однозначно соответствуют друг другу. Например:
Было бы, конечно, удобно, если бы нам удалось аккуратно «вытянуть» одну нить ДНК и спокойно, нуклеотид за нуклеотидом, «прочесть» эту нить от начала до конца. При таком, идеальном, методе секвенирования (чтения ДНК) никаких хитрых алгоритмов не понадобилось бы. К сожалению, на данном этапе такое невозможно, и приходится довольствоваться результатами того секвенирования, которое есть.
Что такое секвенирование?
Секвенирование (sequencing) – это общее название методов, которые позволяют установить последовательность нуклеотидов в молекуле ДНК. В настоящее время нет ни одного метода секвенирования, который бы работал для молекулы ДНК целиком; все они устроены так: сначала готовится большое число небольших участков ДНК (клонируется молекула ДНК многократно и «разрезается» в случайных местах), а потом читается каждый участок по отдельности.
Клонирование происходит либо просто выращиванием клеток в чашке Петри, либо (в случаях, когда это было бы слишком медленно или по каким-то причинам не получилось бы) при помощи так называемой полимеразной цепной реакции. В кратком и неточном изложении работает она примерно так: сначала ДНК денатурируют, т.е. разрушают водородные связи, получая отдельные нити. Затем к ДНК присоединяют так называемые праймеры; это короткие участки ДНК, к которым может присоединиться ДНК-полимераза – соединение, которое, собственно, и занимается копированием (репликацией) нити ДНК.

Рисунок из Википедии
На следующем этапе полимераза копирует ДНК, после чего процесс можно повторять: после новой денатурации отдельных нитей будет уже вдвое больше, на третьем цикле – вчетверо, и так далее.
Все эти эффекты достигаются в основном с помощью изменений температуры смеси из ДНК, праймеров и полимеразы; для наших целей важно, что это достаточно точный процесс, и ошибки в нём редки, а на выходе получается большое число копий участков одной и той же ДНК. Разные методы секвенирования отличаются друг от друга не методами клонирования, а тем, как потом прочесть получившийся «суп» из многочисленных копий одной и той же ДНК.
Секвенирование по Сэнгеру
Первым методом секвенирования, который учёные сумели применить для обработки целых геномов (в том числе генома человека), стало секвенирование по Сэнгеру (Sanger sequencing). Смысл таков: участок ДНК клонируется, после чего полученная смесь делится на четыре части. Каждая часть помещается в активную среду, где присутствуют:
Собственно, процесс практически идентичен клонированию ДНК, с которым мы встретились в предыдущем разделе. Разница только в том, что теперь в один из нуклеотидов подмешаны «ложные» нуклеотиды; они могут образовать точно такую же водородную связь, но не могут продолжить свою нить дальше.
В результате в каждой части образуется большое число копий префиксов исследуемого участка ДНК, которые имеют разную длину, но всегда заканчиваются на одну и ту же букву – в зависимости от того, когда повезёт взять в процесс клонирования «ложный» нуклеотид. Например, в пробирке, где все последовательности заканчиваются на Т, из нашего примера выше получилась бы смесь из следующих префиксов:
ATGCAGAACAGACGATCAGCGACACTTTA (образец)
AT
ATGCAGAACAGACGAT
ATGCAGAACAGACGATCAGCGACACT
ATGCAGAACAGACGATCAGCGACACTT
ATGCAGAACAGACGATCAGCGACACTTT
Как теперь, получив такую смесь, «прочесть» геномную последовательность? Заметим, что в сумме в четырёх пробирках мы получили все возможные префиксы интересующего нас участка. Это значит, что если мы сможем просто измерить длину каждого префикса (точнее говоря, даже не измерить, а просто упорядочить, узнав, кто из них длиннее), то мы сможем узнать и последовательность тоже. Предположим, что мы увидели, что в пробирках лежат префиксы вот такой длины (по порядку, от самого лёгкого 1 до самого тяжёлого 10):A C G T
1, 5, 7, 8, 10 4, 9 3, 6 2
Очевидно, что эта последовательность начинается с А (т.к. самый лёгкий префикс, из одной буквы, заканчивается на A); дальше идёт C, дальше опять A, и так далее. В результате можно прочесть исходный участок: ATGCAGAACA.
А чтобы измерить длину, можно, например, измерить массу всех префиксов во всех пробирках. Чтобы измерить массу, можно, например (разные секвенаторы использовали разные процедуры, но суть от этого не меняется), ионизировать эти молекулы и отправить их наперегонки к заряженному электроду в специальном геле, который создаст трение и замедлит продвижение молекул – этот метод называется электрофорезом. При одинаковом заряде более тяжёлые молекулы будут двигаться медленнее, и в результате получится примерно такая картинка.

Рисунок из Википедии
Видно, что (в идеальном случае) можно просто прочесть последовательность нуклеотидов от самого лёгкого префикса (т.е. префикса из одной буквы) к самому тяжёлому.
Результаты и ошибки сэнгеровского секвенирования
На выходе из сэнгеровского секвенатора получаются короткие участки ДНК, так называемые риды (reads). Для биоинформатики принципиальны две вещи: во-первых, какой длины получаются риды, во-вторых, какие в них могут быть ошибки и как часто (разумеется, на свете нет ничего идеального).
Сэнгеровские риды по этим критериям очень хороши: получаются риды длиной около тысячи нуклеотидов, причём качество начинает заметно падать только после 700-800 нуклеотидов. Сам процесс секвенирования по Сэнгеру, с которым мы познакомились в предыдущем разделе, предопределяет и эффект падения качества (труднее отличить молекулу массой 700 от молекулы массой 701, чем массу 5 от массы 6), и другой неприятный эффект – если в геноме встречается длинная последовательность из одной и той же буквы (…AAAAAAAA…), трудно бывает точно определить, какой она длины – все промежуточные массы попадут в одну и ту же пробирку, некоторые из них могут не встретиться, некоторые слиться друг с другом и т.д. Но всё же сэнгеровское секвенирование даёт отличные результаты с достаточно длинными ридами, которые потом относительно легко собирать. О том, как это делается, мы будем говорить в последующих текстах.
Именно при помощи сэнгеровского секвенирования был впервые расшифрован геном человека. Секвенирование по Сэнгеру применяется и сегодня, но его всё активнее вытесняют другие методы, и применяется оно всё реже. Кому же и почему оно уступило свои позиции?
Секвенаторы второго поколения: Illumina
Современные секвенаторы – это так называемые секвенаторы второго поколения (SGS, second generation sequencing). В них участки ДНК по-прежнему многократно клонируются, но процесс чтения устроен не так, как у Сэнгера. Существует много разных методов, отличающихся довольно существенно, поэтому мы рассмотрим только один из них, один из самых популярных на сегодня – секвенирование по методу Solexa (ныне Illumina; в смене названия не нужно искать глубокий смысл, просто одна компания купила другую).
Процесс секвенирования Illumina проиллюстрирован на рисунке; кроме того, можно посмотреть один из нескольких существующих видеороликов с анимацией этого процесса – в данном случае, действительно, лучше один раз увидеть, чем сто раз прочесть текст. Однако краткие комментарии тоже пригодятся; вот как происходит процесс секвенирования по методу Illumina.
В результате на каждом цикле мы прочитываем одновременно очень большое число нуклеотидов из разных последовательностей. Но за это приходится платить тем, что участки ДНК, которые мы можем прочесть, оказываются гораздо короче, чем в случае секвенирования по Сэнгеру – риды Illumina обычно получаются длиной около 100 нуклеотидов.
Парные риды и постановка задачи
Есть ещё одна важная деталь. Участки ДНК «присасываются» к подложке обоими концами, причём мы можем узнать, какие последовательности соответствуют одному и тому же участку. Это значит, что в реальности мы читаем один и тот же участок, длина которого нам приблизительно известна, сразу с двух сторон. В результате данные получаются примерно такого вида:
причём расстояние между известными строчками (число вопросительных знаков) известно не совсем точно. В зависимости от технологии, можно получить как очень длинные неизвестные фрагменты (около 1000 нуклеотидов), «обрамлённые» двумя ридами длины 100, так и короткие фрагменты, в которых неизвестны буквально два-три десятка нуклеотидов между ридами. И те, и другие могут очень помочь в сборке, и об этом мы тоже будем говорить в следующих сериях.
Итак, теперь мы можем формально поставить задачу сборки геномов. Она звучит так: по большому числу подстрок небольшой длины восстановить исходную длинную строку в алфавите из букв A, C, G, T. В случае секвенирования по методу Illumina – по большому числу пар коротких подстрок, разделённых в исходной строке приблизительно известным расстоянием. Поставив эту задачу, мы можем забыть про биологию, химию и медицину – перед нами чисто алгоритмическая задача. Однако, прежде чем перейти к математике, сделаем ещё несколько замечаний.
Ошибки и показатели качества в секвенаторах второго поколения
Как мы уже знаем, секвенирование всегда содержит ошибки. В секвенаторах Illumina и аналогичных ошибки, как правило, происходят на фазе, когда нужно распознать помеченные нуклеотиды, т.е. понять, каким цветом и с какой силой светятся кластеры из многократно клонированных участков ДНК. На рисунке – типичный пример такой фотографии, порождённой секвенатором Illumina.

Рисунок с сайта medicine.yale.edu
Проблема здесь заключается в том, что из-за неидеальности остальных этапов процесса кластеры никогда не светятся только одним цветом; это всегда смесь всех четырёх цветов с той или иной интенсивностью. Нужно выделить наиболее интенсивную компоненту и оценить, насколько вероятна ошибка в этой букве; эта задача называется base calling (распознавание нуклеотидов). Base calling – это целая наука, в подробности которой мы сейчас вдаваться не будем.
Для нас сейчас важно, что в результате каждому нуклеотиду каждого рида секвенатор ставит в соответствие вероятность того, что этот нуклеотид был распознан правильно. Эти вероятности тоже можно использовать при сборке, и секвенаторы выдают их вместе с собственно ридами.
В итоге типичный рид в так называемом fastq-формате, стандартном для секвенаторов второго поколения, выглядит примерно так:
@EAS20_8_6_1_3_25/1
GCAAAAAACTTACCCCGGAACAGGCCGAGCAGATCAAAACGCTACTGCAATACAGACCATCAAGCACCAACTCCCNNNCGTAGNNNNNNTATGTTNNNNG
+EAS20_8_6_1_3_25/1
HHHHHHHGHHHHHHHHHHHHHHHHHHHHEHHHHHHHHEGHHHHGHHGHEFD?A=A&FFBB>&::===@&@E@E>A#########################
Первая и третья строки содержат имя рида; вторая строка – сама последовательность нуклеотидов. Обратим внимание, что среди букв A, C, G, T встречаются и буквы N – это значит, что секвенатор не смог однозначно определить, какой здесь был нуклеотид, и сдался. А четвёртая строка кодирует, в логарифмическом масштабе, вероятности того, что тот или иной нуклеотид распознан правильно; например, H здесь соответствует вероятности ошибки около одной десятитысячной. Как правило, качество ухудшается к концу рида; в нашем примере, как видите, хвост рида и вовсе не удалось сколь-нибудь надёжно прочитать.
Другие методы секвенирования
Хотя мы подробнее всего рассмотрели секвенатор Illumina (Solexa), на самом деле на этом методе свет клином не сошёлся. Есть и другие секвенаторы второго поколения, с другими свойствами.
В секвенировании лигированием (sequencing by ligation) на фазе, когда уже нужно распознавать нуклеотиды, используют не ДНК-полимеразу и процесс репликации, а специальные короткие «зонды», которые присоединяются к комплементарным нуклеотидам, фиксируются, затем вымываются, и процесс повторяется снова. Так устроен секвенатор SOLiD от Applied Biosystems.
Пиросеквенирование (pyrosequencing) основано на хемилюминесцентных сигналах, которые подают специально модифицированные нуклеотиды, когда соединяются с комплементарным нуклеотидом в прочитываемой нити ДНК; на этом принципе работает, например, секвенатор 454 от 454 Life Sciences.
Принцип работы секвенатора PacBio (от Pacific Biosciences) очень похож на принцип работы Illumina, но у него по-другому устроен метод детектирования – специальные «решётки» позволяют уловить сигналы от отдельных молекул (метод получил название SMRT, single molecule real time sequencing). Это позволяет ускорить процесс, уместить больше ридов на одной подложке (нужно меньше клонировать ДНК, не нужно выращивать большие кластеры) и существенно увеличить длину надёжно прочитываемых ридов.
Недавно появившийся метод ионного полупроводникового секвенирования (на нём основан секвенатор IonTorrent) вместо всего этого просто детектирует соединения (ионы), которые выделяются при присоединении нового нуклеотида к нити ДНК. Это позволяет радикально сократить время и стоимость получаемых ридов, хотя процент ошибок становится больше, и больше становится ошибок в фрагментах из повторяющейся одной буквы.
Человеческая мысль не стоит на месте: методы секвенирования постоянно улучшаются. Однако практически все современные методы выдают относительно короткие риды, от 100 до 400 нуклеотидов; в этом цикле мы будем в основном говорить о том, как собирать именно короткие риды.
Sanger или Illumina?
Человеческий геном был впервые собран на сэнгеровских секвенаторах, причём алгоритмическая сторона того проекта была проработана гораздо меньше, чем сейчас, десять лет спустя. Алгоритмы, которыми собирали первый человеческий геном, значительно проще тех, о которых мы будем говорить в дальнейшем. Однако первый геном всё-таки собрали; может быть, весь алгоритмический прогресс – это никому не нужный миф, и вполне хватило бы старых программ?
Невероятно, но факт: «старые» секвенаторы (первого поколения, сэнгеровские) выдают значительно более подходящие для сборки данные, чем «новые» (второго поколения). Это в основном выражается в длине ридов (reads), тех участков ДНК, которые удаётся последовательно прочесть, и которые, собственно, и нужно собрать в одну большую строчку. Секвенаторы первого поколения выдавали риды длиной более пятисот нуклеотидов, обычно около тысячи. Современные секвенаторы выдают пары ридов, каждый из которых имеет длину около ста нуклеотидов.
На таком уровне становится важной и цена алгоритмической стороны вопроса. Чтобы сборка геномов не занимала дольше и не стоила дороже, чем само их секвенирование, нужно разработать очень быстрые алгоритмы для решения задачи сборки. Об этом пойдет речь в следующей статье.