Синдром Марфана
Рубрика МКБ-10: Q87.4
Содержание
Определение и общие сведения [ править ]
Распространенность оценивается в 1/5000, оба пола в равной степени подвержены заболеванию. Наследуется аутосомно-доминантно.
Этиология и патогенез [ править ]
В подавляющем большинстве случаев синдром Марфана вызван мутациями гена FBN1 (15q21), который кодирует белок фибриллин-1, необходимый для формирования соединительных тканей. Были идентифицированы также пограничные формы синдрома Марфана, которые являются вторичными по отношению к мутациям в гене TGFBR2, расположенный на хромосоме 3, который кодирует рецептор TGF-бета.
Клинические проявления [ править ]
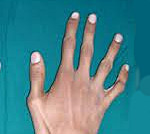
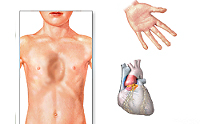
Особенности внешнего вида: арахнодактилия (длинные и тонкие фаланги пальцев рук и ног), удлинение самих конечностей, слабость связочного аппарата, куриная грудь, искривление позвоночника, долихоцефалический череп. Бывают пороки сердца, аневризма аорты, тромбозы вен. В некоторых случаях выявляют тяжелые эндокринные нарушения и нервно-психические расстройства. Со стороны глаз наблюдают вывихи хрусталика в переднюю камеру или стекловидное тело, в результате может развиться вторичная глаукома.
При синдроме Марфана при подвывихе хрусталик смещается вверх, бывают также микрофакия, колобома хрусталика.
Отмечают иридодонез, миоз, могут быть мегалокорнеа или микрокорнеа, высокая степень близорукости, атрофия зрительного нерва. Встречаются случаи пигментной дегенерации сетчатки.
Синдром Марфана: Диагностика [ править ]
Условия диагностики синдрома Марфана:
• при отсутствии пораженного родственника (мать, отец, сибс, ребенок) и негативных ДНК тестов (FBN1) должен присутствовать главный признак в двух различных системах и один малый признак из третьей системы;
• при выявлении мутации гена FBN1 достаточно одного главного критерия любой системы и вовлечения еще одной системы.
Диагностические признаки следующие.
Легочная система считается вовлеченной при наличии одного малого признака.
• Кожа и наружные покровы:
Система считается вовлеченной при наличии одного главного или одного малого критерия.
• Твердая мозговая оболочка:
Рентгенологическое исследование и КТ.
Показания к консультации других специалистов
Все больные с синдромом Марфана должны наблюдаться у кардиолога, офтальмолога и ортопеда.
Наиболее важные аспекты этой патологии для неонатологов и хирургов:
• затруднение интубации из-за подвижности височнонижнечелюстного сустава и шейного отдела позвоночника;
• опасность внезапного повышения или снижения АД во время операции;
• осторожное применение мышечных релаксантов при миопатическом синдроме (возможен парадоксальный или пролангированный эффект);
• возможность летальной желудочковой аритмии и бактериального эндокардита в послеоперационном периоде при пролапсе клапанов сердца с регургитацией;
• разрыв аневризмы аорты;
• повышенный риск спонтанного пневмоторакса (5%);
• повышенная частота пневмоний и хронических эмфиземоподобных изменений;
• снижение жизненной емкости легких, увеличивающее риск анестезиологических осложнений.
Нестабильность шейного отдела позвоночника изредка создает проблемы, что необходимо учитывать анестезиологам при проведении эндотрахеальной интубации, но истинная частота встречаемости этой патологии неизвестна.
Дифференциальный диагноз [ править ]
Дифференциальный диагноз включает MASS-синдром, синдром Шпринтцена-Гольдберга, пролапс митрального клапана, синдром Элерса-Данло и другие заболевания, которые сопровождаются аневризмой аорты, например, синдром Лойса-Дица.
Синдром Марфана: Лечение [ править ]
У большинства больных деформация позвоночника развивается постепенно в течение детского периода. При наличии кифоза и сколиоза умеренной тяжести показано консервативное лечение.
Прием β-адреноблокаторов (пропранолол) в целях профилактики аневризмы аорты.
Патологическая деформация позвоночника может возникнуть на любом уровне и в любом возрасте. Иногда новорожденные имеют тяжелую деформацию, требующую инструментального вмешательства. Опасность раннего хирургического лечения заключается в разрушении зон роста позвонков и снижении высоты грудной клетки.
При сколиотической деформации, превышающей 40°, показана хирургическая стабилизация позвоночника.
Профилактика [ править ]
Прочее [ править ]
Синонимы: синдром аневризмы аорты в связи с аномалиями TGF-бета-рецепторов
Синдром Лойса-Дитца это редкое генетическое заболевание соединительной ткани, которое характеризуется широким спектром краниофациальных, скелетных и сосудистых проявлений.
Синдром Лойса-Дитца имеет четыре генетических подтипа, наследуется аутосомно-доминантно.
Синдромы врожденных аномалий, вовлекающих преимущественно конечности

14-16 октября, Алматы, «Атакент»
150 участников из 10 стран. Новинки рынка стоматологии. Цены от производителей
Общая информация
Краткое описание
Протокол «Синдромы врожденных аномалий, вовлекающих преимущественно конечности»
Код по МКБ:
— Отсуствия (недорозвития) ногтей, надколенника
— Сиреномиелии (сращение нижних конечностей)
— Тромбоцитопения с отсутствием лучевой кости (TAR-синдром)
Q87.4 синдром Марфана

Автоматизация клиники: быстро и недорого!
— Подключено 300 клиник из 4 стран

Автоматизация клиники: быстро и недорого!
Мне интересно! Свяжитесь со мной
Классификация
1. Умственная отсталость.
2. Легкая умственная отсталость.
3. Умеренная умственная отсталость.
4. Тяжелая умственная отсталость.
5. Глубокая умственная отсталость.
Диагностика
Диагностические критерии
Жалобы и анамнез: задержка в психоречевом развитии, снижение мышления, памяти, внимания, расторможенность, врожденные пороки развития конечностей.
Физикальное обследование
Лабораторные исследования: общий анализ крови и мочи.
Инструментальные исследования:
1. Электроэнцефалография (ЭЭГ): на ЭЭГ задержка формирования возрастной корковой ритмики, диффузные изменения электрогенеза.
2. Электромиография (ЭМГ).
3. Компьютерная томография головного мозга (КТ).
Показания для консультации специалистов:
Синдром Марфана
МКБ-10
Общие сведения
Причины синдрома Марфана
Синдром Марфана относится к врожденным аномалиям, наследуемым по аутосомно-доминантному типу, с выраженным плейотропизмом, варьирующей экспрессивностью и высокой пенетрантностью. В основе синдрома Марфана лежат мутации в гене FBN1, отвечающем за синтез фибриллина – важнейшего структурного белка межклеточного матрикса, придающего эластичность и сократимость соединительной ткани. Аномалия и дефицит фибриллина при синдроме Марфана приводят к нарушению формирования волокнистых структур, потере прочности и упругости соединительной ткани, невозможности выдерживать физиологические нагрузки. Гистологическим изменениям в большей степени подвержены стенки сосудов эластического типа и связочный аппарат (в первую очередь, аорта и цинновая связка глаза, содержащие наибольшее количество фибриллина).
Классификация синдрома Марфана
В зависимости от количества пораженных систем выделяют несколько форм синдрома Марфана:
Степень тяжести изменений при синдроме Марфана может быть легкой, средней и тяжелой. По характеру течения дифференцируют прогрессирующий и стабильный синдром Марфана.
Симптомы синдрома Марфана
Синдром Марфана характеризуется сочетанным поражением скелета, глаз, сердечно-сосудистой и нервной систем; многообразием проявлений, варьированием сроков появления первых признаков заболевания; хроническим прогредиентным течением.
При синдроме Марфана отмечаются нарушение функции суставов (гипермобильность); деформация грудной клетки (воронкообразная или килевидная форма), деформация позвоночника (сколиоз, кифоз, кифосколиоз, подвывихи и вывихи шейного отдела, спондилолистез), а также плоскостопие и протрузия вертлужной впадины.
Самая неблагоприятная неонатальная форма синдрома Марфана проявляется в классическом варианте уже при рождении, приводит к прогрессирующей сердечной недостаточности и летальному исходу на первом году жизни ребенка.
Для большинства случаев синдрома Марфана характерна патология органа зрения, включающая близорукость, вывих/подвывих (эктопию) хрусталика, уплощение и увеличение размера роговицы, гипоплазию радужной оболочки и цилиарной мышцы, косоглазие, изменение калибра сосудов сетчатки. Эктопия хрусталика при синдроме Марфана имеет двухсторонний характер, часто развивается в возрасте до 4-х лет и устойчиво прогрессирует, ухудшая зрительную функцию.
При синдроме Марфана наблюдается поражение других систем и органов: нервной (эктазия твердой мозговой оболочки, в т. ч. пояснично-крестцовое менингоцеле), бронхолегочной (спонтанный пневмоторакс, эмфизема легких, дыхательная недостаточность), кожи и мягких тканей (атрофические стрии), рецидивирующие паховые и бедренные грыжи, вывихи и разрывы связок, а также эктопия почек, опущение мочевого пузыря и матки, варикозное расширение вен и др.
Характерный для синдрома Марфана высокий выброс адреналина может способствовать постоянному нервному возбуждению, гиперактивности, а иногда развитию неординарных способностей и умственной одаренности.
Диагностика
Диагноз синдрома Марфана основывается на семейном анамнезе, наличии у больного типичных диагностических признаков по результатам физикального осмотра, ЭКГ и ЭхоКГ, офтальмологического и рентгенологического обследования, молекулярно-генетического анализа и лабораторных исследований.
За диагностические критерии синдрома Марфана берутся характерные изменения в различных системах и органах; главными (большими) из них считаются: дилатация корня/расслоение восходящей части аорты, эктопия хрусталика и эктазия твердой мозговой оболочки; килевидная/воронкообразная деформация грудной клетки, требующая хирургического лечения; отношение длины верхнего сегмента тела к нижнему 1,05; сколиоз (> 20˚) или спондилолистез; ограничение разгибания в локтевом суставе (
Также применяются фенотипические диагностические тесты, определяющие соотношение кисть/рост (при синдроме Марфана > 11%); длину среднего пальца (> 10 см); индекс телосложения Варги – (масса тела, г/(рост, см)x2 – возраст, годы/100, должно быть
Аортография показана при подозрении на аневризму и расслоение аорты. Наличие эктопии хрусталика уточняют с помощью биомикроскопии и офтальмоскопии; протрузию вертлужной впадины устанавливают методом рентгенографии тазобедренных суставов; эктазию твердой мозговой оболочки – МРТ позвоночника.
При синдроме Марфана определяется возрастание (в 2 раза и более) почечной экскреции метаболитов соединительной ткани: глюкозоаминогликанов и их фракций. Метод прямого автоматического секвенирования ДНК позволяет провести генетическую идентификацию мутаций в гене FBN1.
Необходима дифференциальная диагностика с заболеваниями, внешне напоминающими синдром Марфана: гомоцистинурией, врожденной контрактурной арахнодактилией (синдромом Билса), наследственной артроофтальмопатией (синдромом Стиклера), MASS-синдромом, синдромами Элерса-Данлоса, Лойса-Дитца, Шпринцена–Голдберга, семейной эктопией хрусталика и др.
Лечение синдрома Марфана
Лечение и дальнейшее наблюдение пациентов с синдромом Марфана должно осуществляться группой специалистов: офтальмологом, кардиологом, кардиохирургом, ортопедом, генетиком, терапевтом.
Лечение больных с синдромом Марфана направлено на профилактику прогрессирования заболевания и развития осложнений, в первую очередь в сердечно-сосудистой системе. При диаметре аорты до 4 см назначаются β-адреноблокаторы, антагонисты кальция или ингибиторы АПФ. Хирургическое лечение проводится при недостаточности клапанов сердца, пролапсе митрального клапана, значительном расширении (>5 см) восходящей части и расслоении аорты. Реконструктивные операции на аорте при синдроме Марфана, имеют высокий процент послеоперационной 5-ти и 10-ти летней выживаемости. При необходимости выполняют протезирование митрального клапана. У беременных с синдромом Марфана и выраженной сердечно-сосудистой патологией проводят досрочное оперативное родоразрешение путем кесарева сечения. С целью профилактика инфекционного эндокардита и тромбозов после операционных вмешательств назначаются антибиотики и антикоагулянты.
При синдроме Марфана проводится коррекция зрения с помощью подбора очков и контактных линз, при необходимости – лазерное или хирургическое лечение катаракты, глаукомы, удаление смещенного хрусталика с имплантацией искусственного. При выраженных скелетных нарушениях может потребоваться хирургическая стабилизация позвоночника, торакопластика, эндопротезирование тазобедренных суставов. Применяются также патогенетическая коллагеннормализующая терапия, метаболическая и витаминотерапия.
Прогноз
Прогноз жизни больных с синдромом Марфана определяется, в первую очередь, степенью сердечно-сосудистых изменений, а также поражений скелета и глаз. Имеется высокий риск осложненного течения, снижения продолжительности жизни (90-95% не доживают до 40-50 лет) и внезапной смерти. Своевременная кардиохирургическая коррекция при синдроме Марфана позволяет значительно увеличить продолжительность (до 60-70 лет) и улучшить качество жизни больных.
Больные синдромом Марфана должны находиться под постоянным врачебным наблюдением и регулярно проходить диагностическое обследование. При синдроме Марфана показан низкий или средний уровень физической активности, исключающий занятия контактными видами спорта, спортивные соревнования, изометрические нагрузки, подводное плавание. Женщинам детородного возраста с синдромом Марфана необходимо пройти медико-генетическое консультирование.
Синдром Марфана
Синдром Марфана (СМ), или Марфана-Ашара – это наследственное заболевание соединительной ткани с преимущественным поражением сердечнососудистой системы, скелета и органа зрения. Частота СМ в популяции составляет от 1:3000 до 1:15000.
Впервые этот синдром описан французами – в 1896 г., педиатром Антонином Бернардом Марфаном, и в 1902 г. терапевтом Эмилем Шарлем Ашаром.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ.
В настоящее время в различных семьях идентифицировано более 550 мутаций. Среди обнаруженных мутаций в гене FBN1 57% – миссенс мутации, 18% –фреймшифт (сдвиг рамки считывания), 16% – сплайс сайт, и 8% – нонсенс мутации. Чаще всего при классическом СМ имеет место мутация в одном из доменов FBN1 (epidermal growth factor (EGF)-like domain), ответственных за связывание кальция с фибриллином. Вследствие этого «незащищенный» кальцием фибриллин теряет устойчивость к протеазам, что приводит к дестабилизации микрофибрилл и нарушению их функции. Патологические изменения в одном и том же локусе могут обуславливать разнообразные клинические проявления – от стертой формы с поражением одной из систем организма до классической развернутой.
КЛАССИФИКАЦИЯ
1. Стертая: слабо выраженные изменения в одной, двух системах.
а) слабо выраженные изменения в трех системах.
б) выраженные изменения хотя бы в одной системе (ограниченная форма).
в) выраженные изменения в двух, трех системах и более. II. Характер течения: 1. Прогрессирующий.
III. Генетическая характеристика:
1. Семейная форма (тип наследования).
2. Первичная мутация.
IV. Клинические варианты:
1. Болезнь Марфана (присутствие трех классических признаков, семейный характер заболевания).
2. Синдром Марфана (наличие стертых форм с положительными нижеперечисленными диагностическими тестами).
XVII: Врожденные аномалии [пороки развития], деформации и хромосомные нарушения; разделу Q87.: Другие уточненные синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем и имеет код Q87.4.
КЛИНИКА.
Поскольку фибриллин находится в соединительной ткани различных органов, симптоматика СМ многосистемна и разнообразна. При этом наиболее часто наблюдается сочетанное поражение сердечнососудистой системы, скелета и органа зрения. Естественно, тяжесть состояния и прогноз при СМ зависят, прежде всего, от степени поражения сердца и сосудов. Изменения сердечно-сосудистой системы отмечаются у большинства больных. Их основная причина – потеря способности стенок артерий и клапанных структур сердца выдерживать естественные гемодинамические нагрузки.
Наиболее частая сердечная патология при СМ – недостаточность митрального клапана. Обычно наблюдается поражение эластических структур створок и сухожильных нитей клапана с развитием его пролабирования и его недостаточности. Эта дисфункция митрального клапана рано или поздно у многих перерастает в умеренную или тяжелую митральную недостаточность, требующую хирургической коррекции. Реже бывает аортальная и трикуспидальная недостаточность. Стенозы клапанов для СМ не характерны. В связи с наличием у больных клапанных пороков, заболевание часто осложняется инфекционным эндокардитом. Патологические процессы со стороныаорты при СМ. Расширяется корень аорты, ее клапанное кольцо и синус Вальсальвы. Развивающаяся вследствие этого относительная аортальная недостаточность нередко приводит к кардиомегалии и тяжелой левожелудочковой недостаточности. Самым грозным осложнением является развитие расслаивающей аневризмы аорты с внутристеночной гематомой, проявляющееся выраженным болевым синдромом и тяжелыми гемодинамическими нарушениями, сто зачастую явлется причиной смерти больных СМ. Идентичные, но менее выраженные изменения могут быть и в легочной артерии. Так как при СМ сосудистая патология генерализованная, поражается эластическая ткань всех сосудов. Аневризмы могут возникать не только в различных отделах аорты, крупных ветвях легочной артерии, но и в венечных, сонных, лучевых, локтевых, бедренных, мозговых и других сосудах.
СКЕЛЕТ.
Характерным является внешний вид больных: длинные и тонкие конечности с такими же пальцами, длинные, узкие ногти, «птичье лицо» (большой нос и маловыраженный подбородок).
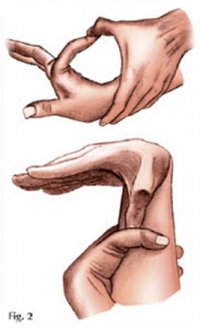
ВЕСЬМА ЧАСТО ПРИ СМ БЫВАЮТ ПОЛОЖИТЕЛЬНЫМИ ТЕСТЫ НА АРАХНОДАКТИЛИЮ:
а) тест большого пальца Steinberg: согнутый 1-й палец выступает за мягкие ткани кисти. При рентгенографии кисти с приведенным большим пальцем его фаланга выступает за скелет метакарпальных костей.
б) тест запястья Walker-Murdoch: При обхватывании запястья другой. При арахнодактилии 1-й и 5-й пальцы соединяются друг с другом.
ОФТАЛЬМОЛОГИЕСКИЕ ПРИЗНАКИ.
Наиболее часто встречается миопия различной степени, гипоплазия радужки, цилиарной мышцы и пигментной каймы зрачкового края, эктопия хрусталиков кверху, внутрь или кнаружи, реже – изменение калибра сосудов сетчатки, катаракта, зрачковая перепонка, косоглазие, дегенерация сетчатки, врожденная или вторичная глаукома. Эктопия хрусталиков вследствие надрывов, разрывов и деструкции связок постоянно прогрессирует, что отражается на зрительных функциях и плохо поддается коррекции очками. Наиболее часто эта патология хрусталиков встречается в среднем школьном возрасте, носит двусторонний характер, но степень ее выраженности может быть различной.
НЕРЕДКО ПРИ СМ НАБЛЮДАЮТСЯ ПОРАЖЕНИЯ СО СТОРОНЫ ДРУГИХ ОРГАНОВ И СИСТЕМ:
Помимо этого, у больных СМ чаще, чем в общей популяции выявляют рецидивирующие паховые и бедренные грыжи, варикозное расширение вен, разрыв межпозвоночных связок, образования межпозвоночных грыж, опущение мочевого пузыря, матки, атрофические изменения кожи, эктазию твердой мозговой оболочки в пояснично-крестцовом отделе и т.д. Последний симптом считается одним из наиболее важных критериев диагностики заболевания.
ДИАГНОСТИКА. ДОПОЛНИТЕЛЬНЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ
1. Лабораторные. Наиболее точным лабораторным признаком СМ является генетическая идентификация мутаций в гене FBN1. + показатели почечной экскреции метаболитов соединительной ткани: оксипролина, оксилизилгликозаминов, гликозаминогликанов и их фракционного состава (увел., как повышенный распад коллагена, а его уровень может определять тяжесть заболевания.
4. Компьютерная томография.
7. Магнитно-резонансная томография (МРТ)
8. Генеалогический анализ. ДИАГНОСТИ
ЛЕЧЕНИЕ.
СИНДРОМ МАРФАНА И БЕРЕМЕННОСТЬ.
Беременность при СМ опасна, по крайней мере, по двум причинам.
1. Имеется риск наследования заболевания, который составляет 50%.
ДИСПАНСЕРНОЕ НАБЛЮДЕНИЕ.
В целях предотвращения прогрессирования заболевания и профилактики осложнений необходимо:
1. Регулярное наблюдение квалифицированных специалистов многопрофильной клиники.
2. Постоянный прием бета-адреноблокаторов (при отсутствии абсолютных противопоказаний).
3. Периодическое выполнение ЭхоКГ, МРТ или КТ для контроля диаметра аорты и клапанных пороков.
4. Профилактика инфекционного эндокардита в течение 6 месяцев после оперативного лечения, а также при имеющихся пороках клапанов.
ПРОГНОЗ.
Продолжительность и качество жизни больных СМ в основном зависит от объема и выраженности поражения сердечно-сосудистой системы, скелета и глаз. Приемлемым для них является низкий или средний уровень физической активности. Из-за риска сердечно-сосудистых осложнений, развития пневмоторакса и возможной дислокации хрусталиков, им не рекомендуется заниматься контактными видами спорта и подводным плаванием. Оперированные пациенты имеют еще больше ограничений, особенно, если принимают антикоагулянты. Раннее начало лечения таких больных позволяет значительно увеличить продолжительность и улучшить качество их жизни. Без лечения средняя продолжительность жизни составляет 32+/-16 лет. При проведении полноценного лечения этот показатель увеличивается до 60 и более лет.
ВЫПОЛНИЛА: СТУДЕНТКА 1 ГР.
1 МЕД. ФАКУЛЬТЕТА
АРАКЕЛЯН Л.В.
ПРЕПОДАВАТЕЛЬ:
ГРЕЧАНИНА Ю.Б
Публикации в СМИ
Синдром Марфана
Синдром Марфана — наследственное заболевание соединительной ткани с вовлечением скелетно-мышечной системы и ССС и патологией глаз. Все доказанные случаи синдрома Марфана — следствие мутации гена фибриллина-1 (#154700, 15q15–q21.3, ген FBN1 [*134797], Â ). Выявлено множество различных мутаций, что объясняет значительную клиническую полиморфность заболевания. Свыше 15% случаев — следствие новых мутаций. Атипичные формы синдрома Марфана могут быть вызваны мутациями в других генах, например в гене трансформирующего фактора роста •• (602091, 14q24, ген LTBP3, Â ). Частота — 1 случай на 10 000–20 000.
Клиническая картина • Скелетно-мышечная система •• Высокий рост •• Астеническое телосложение (длина конечностей не пропорциональна длине туловища) •• Арахнодактилия (длинные тонкие пальцы) •• Деформация грудной клетки •• Высокое арковидное нёбо •• Кифосколиоз •• Слабость связочного аппарата • ССС •• Дилатация корня аорты •• Аортальная регургитация •• Расслаивающая аневризма аорты •• Пролапс митрального клапана •• Регургитация крови при недостаточности митрального клапана • Глаза •• Иридоденез (дрожание хрусталика вследствие слабости цинновой связки) •• Подвывих хрусталика •• Отслойка сетчатки •• Близорукость высокой степени • Другие симптомы (редко) •• Геморрагический синдром •• Спонтанный пневмоторакс.
Дифференциальная диагностика • Гомоцистинурия (236200) — клиника болезни Марфана, умственная отсталость, в моче — гиперэкскреция гомоцистина и метионина • Контрактурная арахнодактилия « Синдром Билса (121050, ген фибриллина-2 FBN2, 5q23–q31, Â ) — разновидность болезни Марфана с нарушением синтеза белка фибриллина-2 • Синдром Элерса–Данло–Русакова (разные типы) • Расслоение стенки артерий c лентигинозом (600459) • Синдром врождённой гиперрастяжимой кожи с марфаноподобным фенотипом (*150240, 7q31.1–q31.3, Â ) • Трисомия 8 имеет костные проявления, как при синдроме Марфана, но без глазных и сердечно-сосудистых признаков • Марфаноидное телосложение с микроцефалией и гломерулонефритом (248760, r ). Клинически: умственная отсталость, микроцефалия, высокое нёбо, прогнатия, марфаноидный внешний вид, гломерулонефрит, почечная недостаточность, расширение IV желудочка.
Лабораторные исследования неспецифичны • Отмечают непостоянную гиперэкскрецию гликозаминогликанов и оксипролина с мочой • Рекомендовано исследование уровня гомоцистина в моче, чтобы исключить гомоцистинурию.
Специальные исследования • Исследование с применением щелевой лампы — слабость цинновой связки, склонность к подвывиху хрусталика • Обзорные рентгенограммы позвоночного столба в период роста — обнаружение и оценка выраженности сколиоза • Ежегодные скрининговые ЭхоКГ, начиная с пубертатного периода, — бессимптомная дилатация корня аорты и дегенерация клапанов.
ЛЕЧЕНИЕ
Тактика ведения. Наблюдение у участкового терапевта, кардиолога, офтальмолога и хирурга-ортопеда (при возможности, генетическое консультирование). Физическую активность следует ограничить.
Хирургическое лечение. Реконструктивная сердечно-сосудистая операция.
Лекарственная терапия • Пропранолол может отсрочить развитие или замедлить прогрессирование расслоения аорты. Дозу подбирают до достижения желаемой ЧСС (в покое — 60 в минуту, при нагрузке — не более 80 в минуту) • Эстрогены в сочетании с прогестероном для стимуляции пропорционального полового созревания у девушек (под наблюдением эндокринолога).
Наблюдение • Частые обследования (по крайней мере 2 р/год) в период роста, особенное внимание слудует обращать на ССС и позвоночник (сколиоз) • При поражении сердца или увеличении диаметра корня аорты более 50 мм необходимо рассмотреть возможность хирургического вмешательства • При наличии подвывиха хрусталика — хирургическая коррекция, но учитывая высокий риск развития глаукомы, операцию нужно предлагать только тем, кто не может обойтись корригирующими линзами.
Осложнения • Септический эндокардит • Расслаивающая аневризма аорты • Недостаточность аортального или митрального клапана • Дилатация сердца • Отслойка сетчатки.
Течение и прогноз. До широкого распространения хирургической коррекции сердечно-сосудистой патологии большинство пациентов с синдромом Марфана умирали до 35 лет. При адекватной коррекции продолжительность жизни большинства пациентов может быть нормальной.
Беременность. Беременных женщин с синдромом Марфана нужно вести как пациенток высокого риска, лучше с участием кардиолога. Результат обычно благоприятный.
МКБ-10 • Q87.4 Синдром Марфана
Приложение. Майгеля синдром (203760, мутации гена коллагена a 2, r ). Марфаноидное телосложение, арахнодактилия.
Код вставки на сайт
Синдром Марфана
Синдром Марфана — наследственное заболевание соединительной ткани с вовлечением скелетно-мышечной системы и ССС и патологией глаз. Все доказанные случаи синдрома Марфана — следствие мутации гена фибриллина-1 (#154700, 15q15–q21.3, ген FBN1 [*134797], Â ). Выявлено множество различных мутаций, что объясняет значительную клиническую полиморфность заболевания. Свыше 15% случаев — следствие новых мутаций. Атипичные формы синдрома Марфана могут быть вызваны мутациями в других генах, например в гене трансформирующего фактора роста •• (602091, 14q24, ген LTBP3, Â ). Частота — 1 случай на 10 000–20 000.
Клиническая картина • Скелетно-мышечная система •• Высокий рост •• Астеническое телосложение (длина конечностей не пропорциональна длине туловища) •• Арахнодактилия (длинные тонкие пальцы) •• Деформация грудной клетки •• Высокое арковидное нёбо •• Кифосколиоз •• Слабость связочного аппарата • ССС •• Дилатация корня аорты •• Аортальная регургитация •• Расслаивающая аневризма аорты •• Пролапс митрального клапана •• Регургитация крови при недостаточности митрального клапана • Глаза •• Иридоденез (дрожание хрусталика вследствие слабости цинновой связки) •• Подвывих хрусталика •• Отслойка сетчатки •• Близорукость высокой степени • Другие симптомы (редко) •• Геморрагический синдром •• Спонтанный пневмоторакс.
Дифференциальная диагностика • Гомоцистинурия (236200) — клиника болезни Марфана, умственная отсталость, в моче — гиперэкскреция гомоцистина и метионина • Контрактурная арахнодактилия « Синдром Билса (121050, ген фибриллина-2 FBN2, 5q23–q31, Â ) — разновидность болезни Марфана с нарушением синтеза белка фибриллина-2 • Синдром Элерса–Данло–Русакова (разные типы) • Расслоение стенки артерий c лентигинозом (600459) • Синдром врождённой гиперрастяжимой кожи с марфаноподобным фенотипом (*150240, 7q31.1–q31.3, Â ) • Трисомия 8 имеет костные проявления, как при синдроме Марфана, но без глазных и сердечно-сосудистых признаков • Марфаноидное телосложение с микроцефалией и гломерулонефритом (248760, r ). Клинически: умственная отсталость, микроцефалия, высокое нёбо, прогнатия, марфаноидный внешний вид, гломерулонефрит, почечная недостаточность, расширение IV желудочка.
Лабораторные исследования неспецифичны • Отмечают непостоянную гиперэкскрецию гликозаминогликанов и оксипролина с мочой • Рекомендовано исследование уровня гомоцистина в моче, чтобы исключить гомоцистинурию.
Специальные исследования • Исследование с применением щелевой лампы — слабость цинновой связки, склонность к подвывиху хрусталика • Обзорные рентгенограммы позвоночного столба в период роста — обнаружение и оценка выраженности сколиоза • Ежегодные скрининговые ЭхоКГ, начиная с пубертатного периода, — бессимптомная дилатация корня аорты и дегенерация клапанов.
ЛЕЧЕНИЕ
Тактика ведения. Наблюдение у участкового терапевта, кардиолога, офтальмолога и хирурга-ортопеда (при возможности, генетическое консультирование). Физическую активность следует ограничить.
Хирургическое лечение. Реконструктивная сердечно-сосудистая операция.
Лекарственная терапия • Пропранолол может отсрочить развитие или замедлить прогрессирование расслоения аорты. Дозу подбирают до достижения желаемой ЧСС (в покое — 60 в минуту, при нагрузке — не более 80 в минуту) • Эстрогены в сочетании с прогестероном для стимуляции пропорционального полового созревания у девушек (под наблюдением эндокринолога).
Наблюдение • Частые обследования (по крайней мере 2 р/год) в период роста, особенное внимание слудует обращать на ССС и позвоночник (сколиоз) • При поражении сердца или увеличении диаметра корня аорты более 50 мм необходимо рассмотреть возможность хирургического вмешательства • При наличии подвывиха хрусталика — хирургическая коррекция, но учитывая высокий риск развития глаукомы, операцию нужно предлагать только тем, кто не может обойтись корригирующими линзами.
Осложнения • Септический эндокардит • Расслаивающая аневризма аорты • Недостаточность аортального или митрального клапана • Дилатация сердца • Отслойка сетчатки.
Течение и прогноз. До широкого распространения хирургической коррекции сердечно-сосудистой патологии большинство пациентов с синдромом Марфана умирали до 35 лет. При адекватной коррекции продолжительность жизни большинства пациентов может быть нормальной.
Беременность. Беременных женщин с синдромом Марфана нужно вести как пациенток высокого риска, лучше с участием кардиолога. Результат обычно благоприятный.
МКБ-10 • Q87.4 Синдром Марфана
Приложение. Майгеля синдром (203760, мутации гена коллагена a 2, r ). Марфаноидное телосложение, арахнодактилия.