Пролиферация лимфоидных элементов что это
Распределение лимфоцитов в коже в зависимости от фенотипа, которое наблюдается в физиологических условиях, находит отражение в развитии лимфопролиферативных заболеваний: Т-лимфопролиферативные опухоли развиваются обычно в верхних слоях дермы и для них характерен экзоцитоз лимфоцитов в эпидермис вплоть до образования внутриэпидермальных микроабсцессов из Т-лимфоцитов (эпидермотропизм). В-лимфопролиферативные процессы возникают в глубоких слоях дермы и обычно не носят эпидермотропный характер. Эти опухоли значительно чаще бывают при системных лимфопролиферативных процессах.
В группе злокачественных лимфом кожи удельный вес заболеваний, в основе которых лежит Т-лимфоцитарная пролиферация, значительно превосходит удельный вес В-лимфом, что отражает реальное соотношение Т- и В-лимфоцитов в нормальной коже. Поданным G. Burg, Т-лимфомы кожи составляют приблизительно 65% всех вариантов ЗЛК, В-лимфомы кожи — 25%, 10% приходится на неклассифицируемые лимфомы.
В нормальной коже лимфоциты в периваскулярных областях представлены почти одинаковым количеством хелперов и супрессоров, хелперносупрессорный индекс обычно равен 0,93—0,96. Встречается также определенное количество клеток, обладающих киллерной активностью (натуральные киллеры).
Т-клеточные лимфомы обычно протекают с преимущественной пролиферацией лимфоцитов, имеющих хелперный фенотип, гораздо реже пролиферируют клетки с супрессорным или киллерным фенотипом.
Преобладание пролиферации Т-лимфоцитов с фенотипом Т-хелперов у больных ЗЛК, возможно, связано с тропностью вирусов типа HTVL-1 к субпопуляции Т-хелперов и способностью этих вирусов к длительной персистенции в таких клетках. Известно, что большая часть Т-лимфом кожи, в том числе наиболее распространенное заболевание этой группы — грибовидный микоз и его лейкемический вариант — синдром Сезари, протекают с преимущественной пролиферацией Т-хелперов, в частности Т2-хелперов-клеток памяти.
Сопоставление клинического течения злокачественных лимфом кожи с принадлежностью клона пролиферирующих злокачественных клеток к определенной субпопуляции Т-лимфоцитов показало, что тяжесть течения заболевания не определяется происхождением злокачественного клона из Т-хелперов, супрессоров или киллеров. Следует учитывать, что фенотип пролиферирующих клеток может меняться. Описаны случаи развития Т-клеточной лимфомы (грибовидного микоза) через год после появления клинически типичной и подтвержденной фе-нотипически В-клеточной лимфомы. Злокачественная Т-лимфома кожи может также предшествовать развитию В-клеточной лимфомы, что объясняют рядом причин: мутагенным действием полихимиотерапии, стимуляцией В-лимфоцитов патологическим клоном Т-лимфоцитов и др. Известны случаи сочетания Т- и В-лимфомы у одного больного, когда поражение кожи протекало по типу Т-лимфопролиферативного процесса, а в периферической крови и регионарных лимфатических узлах обнаруживали атипичные В-лимфоциты.
С развитием молекулярной иммунологии и иммуногенетики появились новые методы, позволяющие с высокой чувствительностью определять клетки злокачественного клона лимфоцитов по маркерам, отражающим перегруппировки или поломки на уровне генных структур клеток (генотипирование клеток). Так, с помощью методов блот-гибридизации по Southern и ПЦР у больных различными клиническими формами ЗЛК удается определять злокачественные клетки, экспрессирующие онкобелки семейства с-тус и c-ras, отражающие нарушение функционирования генов, ответственных за пролиферацию клеток. Кроме того, с помощью ПЦР удается обнаруживать лимфоциты с переустройством структуры Т-клеточного рецептора (TCR), что маркирует перегруппировку гена Т-клеточного рецептора и является признаком злокачественности Т-лимфоцитов. При этом могут определяться изменения структуры как фиксированных цепей TCR (аи(3), так и вариабельных частей этих рецепторов (TCR-V).
Атипичные лимфоциты с перегруппировкой TCR обнаруживают у больных ЗЛК в крови, лимфатических узлах, пораженной коже уже на ранних стадиях заболевания. Значительно чаще определяются лимфоциты с маркерами, отражающими перегруппировку а- и [3- цепей TCR, что обычно сочетается с наличием налим-фоцитах маркеров CD4H и CD45RO+. Гораздо реже встречаются варианты атипичных лимфоцитов с маркерами переустройства у- и 8-цепей TCR (так называемые у-5-Т-клеточные лимфомы). Экспрессия у, 8-рецепторов TCR чаще сочетается с экспрессией CD4+, реже CD8+ маркеров, однако в редких случаях оба эти рецептора (CD4 и CD8) могут не экспрессироваться. Обычно лимфоциты с у- 5-рецепторным комплексом не проявляют тропности к эпидермису. Кожные Т-лимфомы, в основе развития которых лежит пролиферация лимфоцитов с переустройством у- и 5-цепей TCR, обычно характеризуются агрессивным клиническим течением опухолей и плохим прогнозом. При ВЗЛК генотипический анализ выявляет в злокачественных В-лимфоцитах перестройки в генах, кодирующих тяжелые цепи иммуноглобулинов, что также является признаком злокачественности таких клеток.
Диагностика и лечение первичных Т-клеточных лимфом кожи
Первичные Т-клеточные кожные лимфомы представляют собой гетерогенную группу лимфопролиферативных заболеваний, характеризующуюся клональной пролиферацией Т-лимфоцитов в коже. Они составляют 75–80 % всех кожных лимфом. Разграничение первичных лимфом кожи и
Первичные Т-клеточные кожные лимфомы представляют собой гетерогенную группу лимфопролиферативных заболеваний, характеризующуюся клональной пролиферацией Т-лимфоцитов в коже. Они составляют 75–80 % всех кожных лимфом. Разграничение первичных лимфом кожи и вторичных ее поражений при других лимфомах требует тщательного анализа клинической картины, патологических, иммунологических и молекулярно-генетических данных. Первичные лимфомы кожи отличаются от нодальных лимфом характером течения, прогнозом и подходами терапии. Это обстоятельство вызвало необходимость создания подробной классификации, отражающей весь спектр первичных кожных лимфом. В 2005 г. на основе классификации ВОЗ для опухолей гемопоэтической и лимфоидной тканей [1] и классификации лимфом кожи Европейской организации по исследованию и лечению рака (EORTC) [2] создана ВОЗ/EORTC-классификация кожных лимфом, наиболее полно охватывающая весь спектр этих заболеваний [3].
Т- и NK-клеточные лимфомы кожи
— синдром гранулематозной вялой кожи.
— первичная анапластическая крупноклеточная лимфома кожи;
Самыми распространенными подтипами являются: ГМ, синдром Сезари, первичная анапластическая крупноклеточная лимфома кожи и лимфоматоидный папулез. Они составляют приблизительно 95 % всех Т-клеточных кожных лимфом.
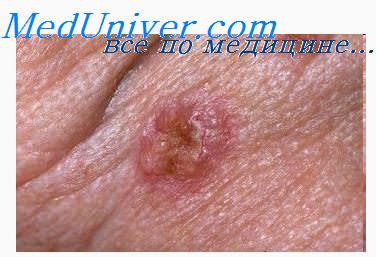
ГМ — первичная эпидермотропная Т-клеточная лимфома кожи, отличительной чертой которой является пролиферация Т-лимфоцитов малых и средних размеров с церебриформными ядрами. Термин «грибовидный микоз» в настоящее время принято использовать только для классического варианта микоза Алибера–Базена, характеризующегося поэтапной эволюцией пятен, бляшек и узлов, или для вариантов со схожим клиническим течением.
Это наиболее часто встречающаяся Т-клеточная опухоль кожи, составляющая 1 % всех неходжкинских лимфом и 50% Т-клеточных кожных лимфом. Средний возраст заболевших — 57 лет, соотношение мужчин и женщин — 2 : 1 [4]. ГМ, особенно на ранних стадиях, может протекать «под маской» различных доброкачественных кожных процессов, таких как хроническая экзема, аллергический контактный дерматит или псориаз. Для начальных кожных проявлений характерна локализация на ягодицах и других защищенных от солнца областях. Заболевание обычно течет благоприятно, медленно прогрессируя в течение нескольких лет или даже десятилетий. Кожные морфологические элементы постепенно эволюционируют от пятен или бляшек до опухолевых узлов с признаками изъязвления. На поздних стадиях заболевания в патологический процесс могут вовлекаться лимфатические узлы и внутренние органы.
Гистологическая картина на ранних стадиях ГМ неспецифична и может быть схожа с таковой при доброкачественном воспалительном дерматозе: отмечаются периваскулярные инфильтраты в сочетании с псориазоформной гиперплазией эпидермиса [5]. Для бляшечной стадии характерен плотный полосовидный инфильтрат в верхней части дермы, содержащий высокий процент церебриформных лимфоцитов с выраженным эпидермотропизмом. Внутриэпидермальные скопления атипичных лимфоцитов (микроабсцессы Потрие) являются характерной чертой этой стадии, но встречаются лишь в 10 % случаев [6]. С прогрессированием в опухолевую стадию эпидермотропизм исчезает, а инфильтрат, состоящий из церебриформных лимфоцитов малых, средних и крупных размеров, становится диффузным и может проникать в подкожную жировую клетчатку.
Прогноз при ГМ напрямую зависит от стадии заболевания, характера и распространенности кожного процесса, а также наличия внекожных поражений.
 |
| Таблица 1. TNMB классификация грибовидного микоза |
В 1978 г. Национальным институтом рака США была предложена TNMB (tumor, node, metastasis, blood) — классификация кожных Т-клеточных лимфом, которая применима для определения стадий ГМ (табл. 1, 2) [8]. Данные о пятилетней выживаемости при грибовидном микозе/синдроме Сезари (ГМ/СС) в зависимости от стадии заболевания следующие: IA — 96 %, IB / IIA — 73 %, IIB / III — 44 %, IV — 27 %.
Кроме классической формы ГМ, имеется несколько вариантов этого заболевания с необычными клиническими и/или гистологическими характеристиками. Из них в классификации ВОЗ/EORTC выделены три клинико-патологических варианта: фолликулотропный ГМ, педжетоидный ретикулез и синдром гранулематозной «вялой» кожи [3].
 |
| Таблица 2. Стадии грибовидного микоза по классификации TNMB (США, 1978) |
Фолликулотропный ГМ характеризуется наличием фолликулотропного и часто неэпидермотропного инфильтрата. Клинически заболевание может проявляться фолликулярными папулами, бляшками и иногда опухолями, локализующимися чаще всего на голове и шее и сопровождающимися алопецией. Гистологически выявляется плотный периаднексальный (перифолликулярный) инфильтрат, состоящий из малых и средних лимфоидных клеток с церебриформными ядрами. Эпидермотропизм может отсутствовать. Волосяные фолликулы часто кистозно расширены, возможна муцинозная дегенерация фолликулярного эпителия.
Педжетоидный ретикулез (моноочаговая форма Ворингера–Колоппа) является благоприятно протекающей формой ГМ и характеризуется наличием одного очага в виде псориазоформной бляшки, локализующейся на нижних конечностях. Гистологическая картина характеризуется акантотическим эпидермисом, содержащим «спонгиоформный» инфильтрат из средних и крупных лимфоидных клеток с вакуолизированной цитоплазмой, расположенных поодиночке или скоплениями.
К одним из самых редко встречающихся вариантов ГМ относится синдром гранулематозной «вялой» кожи, характеризующийся клональной пролиферацией Т-лимфоцитов и дегенерацией эластических волокон в дерме. Заболевание клинически проявляется развитием в крупных складках своеобразных изменений кожи в виде появления складчатых, инфильтрированных и лишенных эластичности образований. Гистологическая картина характеризуется наличием плотного диффузного инфильтрата, состоящего из малых и средних лимфоидных клеток без выраженной атипии ядер, и отсутствием эластических волокон в сосочковой и сетчатой частях дермы. Специфическим гистологическим признаком данного заболевания считается наличие в инфильтрате многоядерных гигантских клеток, имеющих от 20 до 30 концентрически расположенных ядер. В их цитоплазме могут находиться лимфоциты, а также остатки эластических волокон.
Синдром Сезари характеризуется триадой признаков: эритродермия, лимфаденопатия и наличие опухолевых Т-лимфоцитов (клеток Сезари) в коже и периферической крови [9].
Гистологические изменения при синдроме Сезари сходны с таковыми при ГМ, однако эпидермотропизм может быть менее выражен.
Терапия ГМ/СС также зависит от стадии заболевания. На ранних стадиях, при локальных изменениях кожи, эффективна РUVA-терапия, которая позволяет получить полную ремиссию в подавляющем большинстве случаев [11–13]. При I и II стадиях PUVA-терапия может быть использована в комбинации с интерфероном α [14–16]. Применение агрессивных методов лечения (химио- и лучевая терапия) на ранних стадиях не меняет прогноз и, следовательно, неоправдано [17]. При распространенном поражении локальная лучевая терапия, тотальное облучение кожи (ТОК), экстракорпоральный фотоферез позволяют контролировать течение болезни, но имеют ограниченную доступность [18–21]. Когда заболевание становится резистентным к указанным методам лечения, используется комбинированная химиотерапия, однако вне зависимости от ее варианта продолжительность эффекта обычно не превышает 1 года [22–26]. В последнее время в лечении ГМ/СС все большее распространение получает применение биологических препаратов, механизм действия которых основан на специфическом связывании с различными антигенами на мембране опухолевых клеток. К ним относятся интегрированный протеин — Ontak и анти-CD52-моноклональное антитело — алемтузумаб (Кэмпас). Ontak является конъюгатом токсина дифтерии с интерлейкином-2, который после связывания с рецептором к интерлейкину-2 (СD25) подвергается эндоцитозу с последующим высвобождением внутри клетки дифтерийного токсина. Результатом этого процесса является нарушение синтеза белка и в конечном итоге апоптоз Т-лимфоцитов [27]. Кэмпас представляет собой гуманизированное IgG1-моноклональное антитело, специфически связывающееся с CD52-антигеном. Эффекторный механизм Кэмпаса изучен не до конца, он, вероятно, основан на антителозависимой клеточной цитотоксичности [28, 29], комплемент-обусловленном клеточном лизисе [30, 31] и апоптозе [32]. Опухолевые Т-лимфоциты экспрессируют на своей поверхности большое количество молекул CD52 (около 500 000 молекул на лимфоцит), и интенсивность экспрессии CD52 напрямую коррелирует с клиническим эффектом [33, 34]. Основанием для использования Кэмпаса при ГМ является его успешное применение при других Т-клеточных опухолях, например, при Т-клеточном пролимфоцитарном лейкозе [35]. По данным одного из наиболее крупных исследований по использованию Кэмпаса в терапии ГМ/СС, в которое было включено 22 больных со II–IV стадиями заболевания, ранее получавших другие виды лечения, общий ответ на терапию составил 55 %, полная ремиссия достигнута в 32 % случаев. Если предшествующее лечение включало в себя не более двух режимов терапии, общий ответ составлял 80 %. Медиана выживаемости без прогрессии для 12 больных, ответивших на лечение, составила 12 мес [36]. Клиническая картина до и после терапии ГМ препаратом Кэмпас представлена на рис. 1–4.
 |
| Рис. 1. Разрушение зоны эпидермально-дермального стыка. Распространение инфильтрата в эпидермис с образованием абсцессов Потрие. Рис. 2. Единичные лимфоидные клетки среди эпителиоцитов базального слоя эпидермиса (после терапии алемтузумабом) |
Первичная анапластическая лимфома кожи чаще развивается у лиц мужского пола, в основном в возрасте старше 60 лет. Клинически определяются обычно один или несколько узлов (в том числе подкожных), имеющих тенденцию к изъязвлению. Наиболее частая локализация высыпаний — верхние и нижние конечности. Гистологическая картина представлена диффузными инфильтратами в дерме и подкожно-жировой клетчатке, состоящими из клеток с анапластической или иммунобластной морфологией. Эпидермотропизм не характерен. Опухолевые клетки экспрессируют CD30, а также один или несколько пан-Т-клеточных антигенов (CD2, CD3, CD4, CD5). Экспрессия антигена эпителиальных мембран (EMA) и ALK-протеина обнаруживается крайне редко, что отличает первичную кожную анапластическую лимфому от нодальной анапластической лимфомы с поражением кожи [37].
Первичная анапластическая лимфома кожи имеет, как правило, доброкачественное течение. Пятилетняя выживаемость составляет около 90 %. Лучевая терапия или хирургическое удаление применяются в случае единичных очагов. У больных с множественными высыпаниями проводится лечение малыми дозами метотрексата (5–20 мг в неделю) или лучевая терапия. Только быстропрогрессирующее течение заболевания с внекожным распространением требует назначения полихимиотерапии.
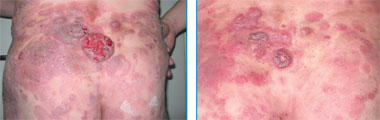 |
| Рис. 3. Множественные пятна, бляшки и опухолевые образования с элементами изъязвления на коже (до начала лечения). Рис. 4. Вид после терапии алемтузумабом |
Тип А характеризуется кожным инфильтратом состоящим из крупных атипичных клеток, напоминающих клетки Рида–Штернберга.
Тип В гистологически напоминает картину ГМ. Анапластические клетки встречаются в малом количестве или отсутствуют.
Тип С характеризуется мономорфным инфильтратом или наличием крупных кластеров CD30 + Т-лимфоцитов. Наиболее эффективным методом лечения является назначение малых доз метотрексата, особенно в случае распространенных высыпаний и частых обострений [3].
Таким образом, диагностика и лечение первичных Т-клеточных кожных лимфом требуют учета многих факторов и использования широкого спектра современных методов обследования (гистологических, иммунологических, молекулярно-генетических), позволяющих правильно определить прогноз и оптимизировать терапию.
По вопросам литературы обращайтесь в редакцию.
В. А. Доронин
ЦКБ № 2 им. Н. А. Семашко, Москва
Пролиферация лимфоидных элементов что это
Миелопролиферативные заболевания включают истинную полицитемию, эссенциальную тромбоцитемию, миелоидную метаплазию и хронический миелолейкоз (ХМЛ). При указанных формах патологии, наряду с интенсивной репликацией клеток-предшественников, сохраняется и их дифференцировка. В связи с этим вначале развития миелопролиферативных заболеваний не возникает цитопении (эритропении, тромбоцитопении, нейтропении).
В соответствии с местом первичного возникновения онкогенной трансформации клеток лимфопролиферативные заболевания делятся на две группы: хронические лимфоидные лейкозы и злокачественные лимфомы, имеющие первоначально внекостномозговую локализацию (лимфатические узлы, селезенка, кожа, лимфоидная ткань слизистой желудка), что отличает их от лейкозов. При лейкозах опухолевый процесс первично развивается в структурах костного мозга.
Хронический миелолейкоз – это опухоль, возникающая из клеток-предшественниц миелопоэза, дифференцирующихся до зрелых форм. Опухолевым процессом поражаются грануло-, моно-, тромбо- и эритроцитарный ростки. Однако безграничный рост в развернутой стадии, как правило, касается только одного ростка – гранулоцитарного.
В 90–97 % случаев хронического миелолейкоза отмечается появление филадельфийской хромосомы (Ph) почти во всех клетках костного мозга – гранулоцитах, моноцитах, эритрокариоцитах, мегакариоцитах. В лимфоцитах ее нет.
Рh-хромосома была найдена в 1960 году в Филадельфии. У больных с хроническим миелолейкозом обнаруживалось специфическое хромосомное нарушение – делеция части длинного плеча у одной из хромосом 21–22-й пары. В такой хромосоме отсутствует приблизительно 40 % генетического материала. Позже было обнаружено, что делетированный участок хромосомы транслоцирован на длинное плечо хромосомы 9. Транслокация может быть и на другие хромосомы.
В настоящее время стало очевидно, что транслокационный вариант мутации, приводящий к появлению филадельфийской хромосомы включает и транслокацию части генетического материала с 9-й хромосомы на 21–22-ю пары хромосом, что приводит к возникновению реципрокной (взаимной) транслокации (9;22) (q34; q11). При этом формируется химерный (слитный) ген BCR-ALB, кодирующий белок р210 с тирозинкиназной активностью.
Заболевание проходит две стадии: доброкачественную (моноклоновую) и злокачественную, терминальную (поликлоновую). Картина крови характеризуется лейкоцитозом со сдвигом до миелоцитов, промиелоцитов и единичных бластов. Кроме омоложения состава гранулоцитов, может быть увеличен процент базофилов или эозинофилов, реже тех и других одновременно (так называемая базофильно-эозинофильная ассоциация).
Красная кровь в моноклоновой стадии болезни существенно не меняется, количество тромбоцитов чаще нормальное. В 20–30 % случаев с самого начала заболевания отмечается тромбоцитоз.
Важнейшим признаком терминальной стадии хронического миелолейкоза является резкое увеличение процента бластных клеток в костном мозге и периферической крови, так называемый бластный криз. Одновременно отмечаются выраженная гранулоцитопения, тромбоцитопения и анемия.
В последние десятилетия выделена прогрессирующая (acselerated) фаза ХМЛ, когда течение заболевания преобретает злокачественный характер. Прогрессирование заболевания может наступить в любой момент, чаще после 3-х лет стабильно текущей формы заболевания. Диагностическими признаками фазы акселерации ХМЛ являются следующие:
1. Миелобласты 10–19 % в крови и/или в костном мозге от всех ядерных клеток
2. Базофилия более 20 %
3. Тромбоцитопения менее 100×109/л или тромбоцитоз более 1000×109/л, не поддающийся терапии
4. Увеличение размера селезенки и лейкоцитоз не поддающийся лечению
5. Дополнительная хромосомная аномалия
6. Возрастающее количество бластов в крови более 15 %
7. Наличие экстрамедулярных очагов кроветворения с пролиферацией бластных клеток
В классификации ВОЗ – онкологическое заболевание лимфоидной ткани, характеризующееся клональной пролиферацией и накоплением длительно-живущих неопластических лимфоцитов (преимущественно СД5+ В клеток) в периферической крови, костном мозге, лимфатических узлах, селезенке, а затем практически во всех внутренних органах и тканях (сердце, легкие, почки, ЖКТ).
Хронический лимфолейкоз – лимфома из малых лимфоцитов, характеризующийся клональной пролиферацией и накоплением предшественников CD+ В-лимфоцитов в периферической крови, костном мозге, лимфатических узлах, почке, а затем во многих других органах и тканях. ХЛЛ – или более частый вид лейкоза, составляет около 25–30 % от всех видов лейкозов.
Значительную часть клеток при хроническом лимфолейкозе составляют В-лимфоциты (80–98 %), но известны и формы болезни с Т-лимфоцитарной пролиферацией.
Этиология ХЛЛ не установлена, в патогенезе важная роль отводится нарушению апоптоза малигнизированных клеток. Факторами пролиферации малигнизированных клеток лимфоидной ткани при ХЛЛ являются такие цитокины, как TNF-α, IL-8, IL-2. Причем, повышение уровня IL-8, усиление экспрессии CD38 – плохие прогностические признаки при В-ХЛЛ.
Специфических хромосомных аномалий при ХЛЛ не выявлено, тем не менее почти у 50 % больных выявляют хромосомные мутации в виде делеции 13q14, у 20 % отмечают делеции 11q22-q23, у 15 % – трисомия 12 и др.
ХЛЛ – болезнь представителей белой расы, преимущественно западного полушария, составляет около 25–30 % всех лейкозов. В восточном полушарии ХЛЛ встречается примерно у 5 % населения.
ХЛЛ – болезнь пациентов преимущественно пожилого возраста, лишь у 10–15 % наличие заболевания диагностируется в возрасте до 50 лет, причем чаще болеют мужчины.
95 % всех случаев ХЛЛ в США и Европе приходится на В-клеточный фенотип.
Установлена определенная роль наследственного фактора в развитии патологии, в то же время очевидно, что воздействие таких факторов внешней среды, как ионизирующая радиация, канцерогенов химической природы, лекарственных препаратов не играет роли инициирующих патогенов в развитии ХЛЛ.
В основе развития ХЛЛ лежит нарушение апоптоза малигнизированных клеток и накопление их преимущественно в G0 – фазе клеточного цикла. В связи с дисбалансом соотношения основных про- и антиапоптотических белков семейства гена bcl2, таких как BAX и BAK (индуцирующих апоптоз), а также BAD, BIK, HBK (антиапоптотических ингибиторов).
Установлено, что продуцируемые модифицированными при ХЛЛ клетками цитокины (TNF-α, IL-8, IL-2) обеспечивают аутокринную и паракринную стимуляции и резистентности малигнизированных лимфоидных клеток.
В настоящее время очевидно наличие 2-х генетических вариантов ХЛЛ в зависимости от происхождения В-клеток, отличающихся по мутационному статусу генов вариабельных участков тяжелых цепей иммуноглобулинов (Vн-генов).
Выделяют: 1) первый вариант ХЛЛ – из нативных В-клеток, не прошедших этап мутации Vн-генов в герминальном центре размножения и 2) второй вариант ХЛЛ, возникающий из В-клеток памяти, подвергшихся соматической мутации Vн- генов.
Первый вариант формирования В-ХЛЛ из нативных клеток имеет плохой прогноз, особенно с экспрессией CD38 и характерными цитогенетическими аномалиями, при этом медиана выживаемости составляет 8 лет.
Медиана выживаемости при втором варианте ХЛЛ составляет около 25 лет. Плохим прогностическим признаком при ХЛЛ первого типа является экспрессия молекул ZAP-70. Около 40–60 % больных ХЛЛ диагностируется в связи с развитием лимфоаденомы и высоким лимфоцитозом. Манифестацией вовлечения костного мозга в патологию являются анемия, тромбоцитопения. Возможна инфильтрация лейкозными клетками различных органов и тканей.
Медиана выживаемости у CD5 + пациентов с ХЛЛ значительно выше, чем у CD5- больных и составляет 97,2.
В зависимости от характера клинического течения ХЛЛ выделяют три основных варианта:
● медленно-текущий ХЛЛ (индолентный)
● ХЛЛ с трансформацией в крупноклеточную лимфому (синдром Рихтера ) или пролимфоцитарный лейкоз.
Медленно-текущий вариант характеризуется стабильным течением с длительным сохранением стадии 0 (1), отсутствием инфекционных осложнений, отсутствием протеина ZAP-70. У 50–70 % больных этой группы обнаруживаются признаки соматических гипермутаций Vн-генов лейкемических В-клеток. При цитогенетическом анализе часто выявляется делеция 13q14, свидетельствующая о положительном прогнозе заболевания.
Картина периферической крови характеризуется лимфоцитозом, который на ранних этапах заболевания выражен умеренно, а на развернутой и терминальных стадиях болезни достигает высоких цифр (80–90 % лимфоцитов). Количество эритроцитов и тромбоцитов в периферической крови долгое время остается в пределах нормы или незначительно снижается. Развернутая и терминальная стадии лимфолейкоза характеризуются анемией и тромбоцитопенией, которые обусловлены аутоиммунным процессом – появлением антител к созревающим клеткам костного мозга или к зрелым элементам крови и костного мозга. Иногда в периферической крови появляется большое количество пролимфоцитов и на этом основании выделяют пролимфоцитарную форму хронического лимфолейкоза.
Прогрессирующее течение ХЛЛ характеризуется быстрой сменой стадий заболевания по К.Rai (1975), гиперлимфоцитозом и диффузной инфильтрацией костного мозга, прогрессирующей лимфоаденопатией, гепато- и спленомегалией, аутоинтоксикацией, гипогаммаглобулинемией, активацией инфекций. Характерно развитие аутоиммунной анемии и тромбоцитопении.
При этой патологии, как правило, отсутствуют мутации Vн-генов иммуноглобулинов на фоне высокой экспрессии ZAP-70.
Плохими прогностическими признаками являются мутации типа делеции 11q22-q23, трисомия 12, дисфункция р 53.
Крупные клетки при синдроме Рихтера свидетельствуют о появлении нового злокачественного клона лимфоцитов. Крупноклеточная лимфома встречается у 2–5 % пациентов, сопровождается клиническими признаками генерализации заболевания.
Развитие пролимфоцитарного ХЛЛ из зрелых клеток отмечают в 5–8 % наблюдений, характеризующиеся более агрессивным течением по сравнению с В-клеточным пролимфоцитарным ХЛЛ, формирующимся de novo.
Волосатоклеточный лейкоз – хроническое В-клеточное пролиферативное заболевание, характеризующееся появлением мононуклеаров с выростами цитоплазмы, характеризующееся прогрессирующим течением с развитием панцитопении, спленомегалии, рецидивирующей инфекции.
Волосатые клетки, экспрессируют CD19, СD26, CD22 и коэкспрессируют CD11c, СD25, CD103 как и В-клетки. при классической форме В-клеточного лейкоза. При вариантной форме отсутствуют антигены СD25, CD103.