Признаки дисмегакариоцитопоэза что это
Из дисмегакариоцитопоэза при миелодиспластических синдромах в крови определяется, как правило, тромбоцитопения, изредка тромбоцитоз. Частота тромбоцитопении при миелодиспластических синдромах варьирует по различным источникам от 28 до 65 %. Как правило, снижение числа тромбоцитов сочетается с другими цитопениями. Изолированная тромбоцитопения обнаруживается в единичных наблюдениях. В нашей серии наблюдений тромбоцитопения установлена у 68,3 % больных: в составе 3-ростковой цитопении — у 47,6 %, только в сочетании с анемией — у 17,1 %, только в сочетании с лейкопенией — у 2,4 %, изолированная тромбоцитопения — у 1,2%. Тромбоцитоз свыше 400*109/л был обнаружен в 2 % случаев.
Как и по данным других авторов, мы наблюдали несколько большую частоту тромбоцитопении при «продвинутых» вариантах, чем при РА и РАКС.
Морфологические аномалии тромбоцитов при миелодиспластических синдромах заключаются в анизоцитозе и гипогрануляции. В крови появляются гигантские формы тромбоцитов, а также их обломки, фрагменты мегакариоцитов. Описана субпопуляция тромбоцитов, характеризующаяся при фазовоконтрастном микроскопическом исследовании особой выпуклой шарообразной формой мембраны клеток.
Как показали собственные данные, средний объем тромбоцитов был уменьшен до 5,0 фл (при норме 7,2—11,0 фл) в случаях с тромбоцитопенией. Во всех наблюдениях был констатирован анизоцитоз тромбоцитов на основании увеличения показателя PDW (ширина распределения тромбоцитов по объему).
При миелодиспластических синдромах (МДС) выявлены значительные функциональные нарушения тромбоцитов вследствие изменения строения мембранных структур и метаболизма ферментов. В плотных гранулах тромбоцитов уменьшено содержание аденозиндифосфата, серотонина и тром-боцитарного фактора 4. Кроме того, констатировано уменьшение адгезии тромбоцитов к стеклу и коллагену, а также снижение активности фактора 3. Патологические формы тромбоцитов продуцируют, кроме нормального тромбоксана В2, также и тромбоксан А2 с низкой биологической активностью. У ряда больных длительность жизни тромбоцитов существенно короче по сравнению с нормальной (0,5— 2,65 дня и 6,5—12 дней соответственно).
В связи с функциональной неполноценностью тромбоцитов геморрагический синдром отмечен у больных с нормальным числом тромбоцитов. Удлинение времени кровотечения может быть связано с нарушением агрегации клеток, выявляемой во многих случаях миелодиспластических синдромов пробой с индукцией экзогенных активаторов тромбоцитов — эпинефрином, коллагеном, ристоцетином, арахидоновой кислотой.
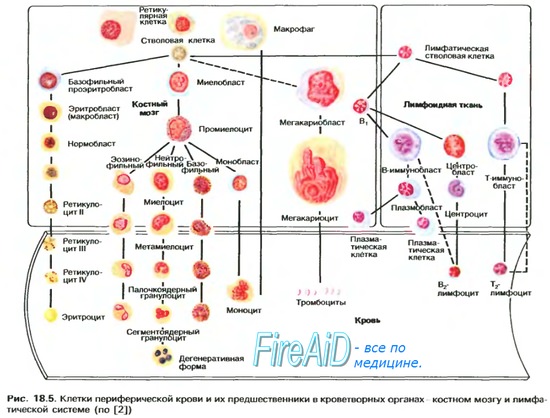
Разнообразные изменения клеток мегакариоцитарного ростка в костном мозге наблюдаются у большинства пациентов. Количество мегакарио-цитов колеблется от единичных клеток до резко повышенных показателей, в том числе в связи с тем, что приблизительная визуальная характеристика мазков не дает точного результата. Подсчет мегакариоцитов в камере также может занизить искомые данные, так как мелкие одноядерные формы неотличимы от клеток гранулоцитарной и моноцитарной линий. Окраска на а-нафтилацетатэстеразу дает возможность визуализировать и идентифицировать на мазках все без исключения патологические формы мегакариоцитов.
Наш опыт свидетельствует о том, что использование цитохимического подхода позволило в отдельных случаях выявить вдвое больше мегакариоцитов, чем при обычной окраске.
Диспластические изменения касаются ядер и цитоплазмы мегакариоцитов. В результате разобщения ядер они располагаются в цитоплазме разъединенно. Вместо многоядерных форм обнаруживаются одно-или двуядерные клетки, а также микроформы мегакариоцитов размером менее 12 мк. Возможно образование мелких мегакариоцитов с компактным ядром, окруженных отложением ретикулина. Цитоплазма клеток либо гипогранулярна, либо содержит аномальные крупные гранулы. Морфологическая аномалия мегакариоцитов определяется в большем числе случаев при РАИБ и РАИБ-Т, чем при РА.
Патологические формы мегакариоцитов сохраняют свою способность отшнуровывать тромбоциты, однако корреляция между числом мегакариоцитов и содержанием тромбоцитов в крови отсутствует. По мнению некоторых исследователей, сочетание двух признаков диспоэза: микрогенераций мегакариоцитов с пельгероидными формами нейтрофилов дает основание для уверенной диагностики миелодиспластического синдрома.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
НОВАЯ КЛАССИФИКАЦИЯ МИЕЛОДИСПЛАСТИЧЕСКИХ (ПРЕДЛЕЙКЕМИЧЕСКИХ) СИНДРОМОВ
Миелодиспластические синдромы (МДС) — группа клональных заболеваний, связанных с поражением гемопоэтической стволовой клетки, харак-теризующихся цитопенией, наличием признаков дисплазии кроветворных кле-ток и повышенным риском развития острых лейкозов. В соответствии с новой классификацией ВОЗ выделяют следующие основные формы МДС: рефрактерные цитопении, рефрактерная анемия с кольцевыми сидеробластами, рефрактерная цитопения с мультилинейной дисплазией, рефрактерная анемия с избытком бластов (РАИБ-1, РАИБ-2), МДС неклассифицируемый, МДС, ассоциированный с del(5q), и рефрактерная анемия детского возраста.
Миелодиспластические синдромы (МДС) — гетерогенная группа клональных заболеваний, в основе возникновения которых лежит поражение гемопоэтической стволовой клетки (ГСК), сопровождающееся неэффективным гемопоэзом, диспластическими изменениями, затрагивающими клетки одной или нескольких линий миелопоэза, повышенным риском развития острых миелоидных лейкозов (ОМЛ).
мические вещества (свинец, мышьяк), противоопухолевые и антивирусные препараты. Термин «миелодиспластические синдромы» (синонимы — «дисмиелодиспластические синдромы», «олигобластный лейкоз») свидетельствует о наличии заболевания кроветворной ткани опухолевой природы, сочетающегося с признаками миелодисплазии.
МДС встречается преимущественно у лиц пожилого и преклонного возраста. Ежегодно заболеваемость в большинстве стран мира составляет 3,0–5,0 на 100 тыс. населения, но возрастает до 20,0 на 100 тыс. у лиц старше 70 лет [1]. Различают МДС, возникающие спонтанно (de novo), и вторичные МДС, развивающиеся в результате применения алкилирующих препаратов и/или лучевой терапии. К числу этиологических факторов, вызывающих возникновение первичных МДС, относят бензол, курение сигарет, воздействие химических веществ, применяющихся в сельском хозяйстве, а также органических растворителей [2].
При МДС могут поражаться все три ростка гемопоэза. Характерным является наличие признаков цитопении при исследовании периферической крови (ПК), сочетающейся с гиперили нормоклеточностью костного мозга (КМ). В качестве основного
патогенетического механизма МДС рассматривается увеличение степени апоптоза активно пролиферирующих ГСК. Процессы программированной гибели ассоциируются с изменениями экспрессии в кроветворных клетках прои антиапоптотических белков. В популяции CD34-положительных бластных клеток КМ у больных с МДС по сравнению с нормой увеличено соотношение продуктов экспрессии онкогенов c-MYC и BCL-2 (соответственно усиливающих апоптоз и повышающих выживаемость клеток) [1].
Важную роль в индукции апоптоза в клетках КМ играет система, включающая антиген Fas/Apo (CD95) и его лиганд Fas-L, которые при иммуноцитохимическом исследовании обнаруживаются на поверхностных мембранах клеток при МДС и не определяются на CD34 + CD14 + клетках в норме. Установлена также корреляция между интенсивностью программированной гибели клеток эритробластического, гранулоцитарного, мегакариоцитарного ряда и стромальных клеток при МДС и экспрессией фактора некроза опухоли α (ФНО-α) и трансформирующего фактора роста β (ТФР-β). С увеличением интенсивности апоптоза при МДС ассоциируется повышенная экспрессия белка р53.
Современная классификация МДС (ВОЗ, 2008) позволяет более четко выделить гомогенные подгруппы больных и имеет большую прогностическую значимость [3]. Она основывается на определении процентного содержания бластов в КМ и ПК, типа и степени диспластических изменений в клетках эритробластического, гранулоцитарного и мегакариоцитарного ряда, данных кариотипирования и молекулярно-генетического анализа (таблица).
При диагностике МДС с целью выявления диспластических изменений должны использоваться идеально приготовленные из свежего материала и хорошо окрашенные мазки. Мазки из проб крови или КМ, подвергавшиеся действию антикоагулянтов на протяжении более двух часов, для этих целей непригодны. Процентное содержание бластов
Данные исследования ПК и КМ при МДС (ВОЗ, 2008)
Рефрактерная цитопения с однолинейной
дисплазией (РЦОД) Рефрактерная анемия (РА);
Рефрактерная нейтропения (РН);
Рефрактерная тромбоцитопения (РТ)
Однолинейная цитопения или бицитопения
Бласты отсутствуют или определяются крайне редко ( 9 /л моноцитов
Дисплазия в ≥ 10% клеток двух или более линий
миелопоэза (нейтрофилы и/или эритроидные предшественники и/или мегакариоциты)
Однолинейная или мультилинейная дисплазия
Отсутствие палочек Ауэра
Рефрактерная анемия с избытком бластов-2
5–9% бластов Палочки Ауэра ±
Однолинейная или мультилинейная дисплазия
10-19% бластов Палочки Ауэра ±
Миелодиспластический синдром некласси-
РЦОД составляет 10-20% всех случаев МДС и чаще встречается у пациентов пожилого возраста (около 65–70 лет), большинством из которых являются больные РА. Однако РН и РТ, наблюдающиеся крайне редко, требуют особого внимания при установлении диагноза. При установлении диагноза РЦОД должны быть исключены проявления дисплазии костномозгового кроветворения неклональной природы, связанные с действием лекарственных препаратов, токсических соединений, с вирусной инфекцией, а также с иммунологическими нарушениями, врожденными заболеваниями, недостаточностью витаминов и т.д.
Рефрактерная анемия. Эритроциты ПК — нормохромные и нормоцитарные, или нормохромные и макроцитарные. Варьирует степень анизоцитоза и пойкилоцитоза. Определяемая при проведении общего анализа крови анемия устойчива к проводимой терапии. Выраженная в умеренной степени лейкопения не сопровождается существенными изменениями в лейкограмме. Морфология нейтрофилов, как правило, не изменена. Лишь в некоторых случаях в ПК определяются агранулярные нейтрофильные палочкои сегментоядерные лейкоциты. Количество тромбоцитов колеблется в пределах нормы, лишь изредка обна-
руживаются гигантские тромбоциты. Бластные клетки в ПК встречаются редко. При этом их количество, как правило, не превышает 1%. Абсолютное количество моноцитов в крови — менее 1,0 х 10 9 /л.
Пунктат КМ у подавляющего большинства больных гиперклеточный. Значительно увеличено количество незрелых ядросодержащих клеток эритробластического ряда. Эритропоэз — нормобластический, макронормобластический или мегалобластический. Эритроидные клетки — с умеренными или выраженными признаками дисплазии. Характерными для дисэритропоэза являются изменения ядер клеток (фрагментация, дольчатость, наличие межъядерных мостиков, кариорексис, многоядерность). В цитоплазме происходят следующие изменения: вакуолизация, диффузное или гранулярное окрашивание при проведении PAS-реакции. Кольцевые сидеробласты, выявляемые при окраске по Перлсу, составляют не более 15% эритроидных клеток-предшественников. Миелобласты составляют менее 5% от общего количества миелокариоцитов. Палочки Ауэра в их цитоплазме не обнаруживаются. При гистологическом исследовании трепанобиоптатов КМ определяются признаки гиперплазии, преимущественно за счет клеток эритробластического ряда. У немногих больных КМ может быть нормоклеточным и даже гипоклеточным.
Различного типа приобретенные клональные цитогенетические аномалии при РА наблюдаются у 50% больных. Они важны для диагностики указанной формы МДС, но не являются специфическими. К числу наиболее часто встречающихся относятся del(20q), +8, аномалии хромосом 5 и/или 7.
Рефрактерная нейтропения. Характеризуется диспластическими изменениями в 10% и более нейтрофилов ПК и КМ, которые проявляются главным образом в наличии гиподольчатых ядер и гипогранулярной цитоплазмы. В клетках других линий миелопоэза значительных проявлений дисплазии не отмечается. Следует исключить вторичную нейтропению, вызванную применением лекарственных средств, воздействием токсических препаратов, обусловленную иммунными механизмами.
зующийся анемией, морфологическими признаками дисплазии в клетках эритробластического ряда и наличием кольцевых сидеробластов (КС) в КМ, количество которых превышает 15%. Выраженных признаков дисплазии в клеточных элементах других линий миелопоэза не отмечается. Содержание миелобластов в КМ ниже 5%, а в ПК они не определяются или их содержание в лейкограмме не превышает 1%. РАКС составляет 3–11% всех случаев МДС. Встречается преимущественно у пожилых людей, с одинаковой частотой у мужчин и женщин. Средний возраст больных составляет 60–73 года.
Клинические проявления обусловлены наличием нормохромной макроцитарной или нормохромной нормоцитарной анемии. У некоторых больных могут дополнительно обнаруживаться признаки тромбоцитопении и нейтропении. В части случаев в ПК может определяться популяция гипохромных ядросодержащих клеток эритробластического ряда с признаками дефектной гемоглобинизации цитоплазмы.
В КМ больных РАКС определяются признаки гиперплазии клеток эритробластического ряда. Признаки дисплазии проявляются в дольчатости ядер клеток эритробластического ряда, появлении клеток с мегалобластоидными признаками. Содержание клеток гранулоцитарного и мегакариоцитарного ряда с диспластическими признаками не достигает 10%. При окраске по Перлсу мазков из стернального пунктата КМ определяется 15% и более КС, содержащих пять и более гранул железа, окружающих не менее одной трети ядра. Нередко обнаруживается значительное количество содержащих гемосидерин макрофагов. При РАКС гранулы железа, которые на ультраструктурном уровне локализуются в митохондриях, определяются и в наименее дифференцированных клетках эритробластического ряда. Клональные хромосомные аномалии в кроветворных клетках выявляются у 5–20% больных РАКС.
Медиана выживаемости больных колеблется в пределах 69–108 мес. Трансформация в острый лейкоз происходит крайне редко (в 1–2% случаев). Рефрактерная цитопения с мультилинейной дисплазией (РЦМД). РЦМД составляет около 30% всех случаев МДС. Это тип МДС, при котором при исследовании ПК у больных, помимо анемии, отмечаются признаки бицитопении или панцитопении. Диспластические изменения обнаруживаются более чем в 10% клеток двух или более линий миелопоэза. Содержание бластных клеток в ПК — менее 1%, а в КМ — менее 5%. Количество моноцитов в ПК менее 1,0 х 10 9 /л. Палочки Ауэра в цитоплазме кроветворных клеток не обнаруживаются. РЦМД диагностируется преимущественно у людей старше 70 лет. Пик заболеваемости у мужчин, болеющих РЦМД несколько чаще, приходится на возраст 70–
74 года, а у женщин — на 75–79 лет.
При исследовании мазков из стернального пунктата, как правило, определяется гиперклеточность КМ. Диспластические изменения в нейтрофилах
проявляются в уменьшении количества гранул в цитоплазме и гипосегментации ядер (псевдопельгеровские лейкоциты). У некоторых больных наблюдается повышенное содержание молодых и незрелых клеток эритробластического ряда. Встречаются эритроидные клетки с ядрами неправильной формы и многодольчатыми, наличием вакуолизированной цитоплазмы. Обнаруживаются мегалобласты. При PAS-реакции в клетках эритробластического ряда отмечается диффузное или гранулярное окрашивание цитоплазмы. Наиболее часто повторяющиеся диспластические изменения в мегакариоцитах — гиподольчатость ядер и наличие микроформ с двудольчатыми ядрами [6].
Почти у 50% больных РЦМД выявляются клональные аномалии хромосом — трисомия 8, моносомия 7, del(7q), моносомия 5, del(5q), del(20q) и более сложные изменения кариотипа [5].
Прогностические факторы обусловлены выраженностью цитопении и дисплазии. Медиана выживаемости больных РЦМД составляет 30 мес. У больных со сложными изменениями кариотипа она сходна с наблюдающейся у больных с РАИБ [3]. Трансформация в острый лейкоз на протяжении первых двух лет заболевания у пациентов с РЦМД составляет до 10%.
При изучении срезов трепанобиоптатов КМ обнаруживаются изменения нормальной гистопографии. Отмечается смещение очагов эритропоэза и мегакариоцитопоэза к паратрабекулярным зонам, которые в норме заняты преимущественно клеточными элементами гранулоцитопоэза. Бласты при РАИБ имеют тенденцию к образованию кластеров, не связанных с костными трабекулами и сосудами. Прежде эти данные гистологического исследования расценивались в качестве аномальной локализации незрелых клеток-предшественников (ALIP). Для идентификации подобных кластеров особенно ценным может быть иммуногистохимическое определение CD34-положительных клеток. Почти у 15% больных с РАИБ при гистологическом исследовании трепанобиоптатов КМ обнаруживаются признаки выраженного фиброза. В этих случаях необходима дифференциальная диагностика с МДС, связанным с терапией, с миелопролиферативными новообразованиями и реактивными состояниями, сопровождающимися рядом других заболеваний (коллагенозы, метастатические поражения КМ, миелопатия при ВИЧ-инфекции и т.д.).
Важную роль в диагностике РАИБ призваны играть результаты иммунофенотипирования. В частности, при использовании методов проточной цитофлуориметрии в ПК и КМ определяется увеличенное количество клеток, экспрессирующих ассоциированные с гемопоэтическими клеткамипредшественниками антигены CD34 и/или CD117. На этих же клетках обычно выявляются антигены CD38, HLA-DR и миелоидно-ассоциированные антигены CD13 и/или CD33. В популяции бластных клеток может обнаруживаться асинхронная экспрессия антигенов, связанных с созреванием клеток гранулоцитарного ряда — CD15, CD11b и/или CD65. В 20% случаев на бластных клетках определяется аберрантная экспрессия антигена CD7 и у 15% больных — CD56. Крайне редко выявляется экспрессия других лимфоидных маркеров [6]. Клональные цитогенетические аномалии, включая
Для установления прогноза заболевания, определения выживаемости больных и частоты трансформации в ОМЛ важное значение имеет выделение 2 основных категорий РАИБ. Определяющим при идентификации РАИБ-1 является наличие до 5% бластов в ПК и 5–9% бластных клеток в КМ. Диагноз РАИБ-2 устанавливается при наличии в КМ 10–19% бластов. К этой же категории должны быть отнесены больные, у которых в КМ определяется менее 10% бластов, но содержание бластных клеток в ПК колеблется в пределах 5–19%. Наличие в бластах палочек Ауэра позволяет автоматически квалифицировать тот или иной случай как РАИБ-2, независимо от процентного содержания бластов в ПК и КМ. Медиана выживаемости больных с РАИБ-1 составляет 16 мес, а пациентов с РАИБ-2 — 9 мес. При наличии палочек Ауэра в цитоплазме клеток медиана выживаемости составляет 12 мес. С плохим прогнозом ассоциируется экспрессия на поверхностных мембранах бластных клеток антигена CD7 [3].
Миелодиспластический синдром с изолированной del(5q). МДС с изолированной делецией хромосомы 5 (5q-синдром) характеризуется наличием анемии с/без других видов цитопении и/или тромбоцитозом. Единственной выявляемой цитогенетической аномалией является del(5q). При 5q-синдроме содержание миелобластов составляет менее 5% всех ядросодержащих клеток КМ и менее 1% клеток ПК. Лейкоциты с палочками Ауэра в цитоплазме отсутствуют. Термин «5q-синдром», как правило, используется для обозначения случаев с наличием макроцитарной анемии, нормальным или повышенным количеством тромбоцитов и гиперплазией клеток эритробластического ряда в КМ.
РАКС, РЦМД, РАИБ. Встречается преимущественно у пожилых людей, но может диагностироваться и у детей. Симптоматика в основном такая же, как и при других формах МДС. Не выявляются и какиелибо специфические изменения цитоморфологических признаков кроветворных клеток.
Диагноз МДС-Н устанавливается в следующих случаях. Во-первых, у больных с рефрактерной цитопенией, затрагивающей один или два ростка гемопоэза, и однолинейной или мультилинейной дисплазией и наличием 1% бластов в ПК. Во-вторых, в случае МДС с однолинейной дисплазией, которая сочетается с панцитопенией. И, наконец, у больных с персистирующей цитопенией(ями) при содержании бластов в ПК менее 1% и в КМ менее 5% и наличии признаков дисплазии менее чем в 10% клеток одной или более линий миелопоэза. При этом должны определяться цитогенетические аномалии, выявляемые при том или ином подтипе МДС. Динамическое клинико-гематологическое наблюдение позволяет проследить эволюцию МДС-Н в один из специфических подтипов заболевания.
Миелодиспластический синдром в детском возрасте. МДС в детском возрасте встречается нечасто, составляя менее 5% всех опухолей кроветворной и лимфоидной тканей у детей младше 14 лет. Ежегодная заболеваемость МДС составляет 0,18 на 100 тыс. детского населения, в то время как частота ОЛЛ — 3,85, а ОМЛ — 0,54. Первичный, или возникающий de novo, МДС следует отличать от «вторичных МДС», обусловленных врожденными или приобретенными нарушениями костномозгового кроветворения, и МДС, связанного с терапией, развивающегося после действия цитотоксических препаратов, которые применяются для лечения детей с опухолями или с неонкологическими заболеваниями. Кроме того, в соответствии с новой классификацией ВОЗ (2008), к рассматриваемой нозологической форме не относится МДС, ассоциированный с синдромом Дауна. Наряду с ОМЛ и транзиторным аномальным миелопоэзом, он включен в новую категорию «Миелоидные пролиферации, связанные с синдромом Дауна».
У детей с МДС отмечаются те же морфологиче-
ские, цитохимические и иммунофенотипические признаки, что и у взрослых больных. В то же время наблюдаются и существенные отличия, особенно у тех пациентов, у которых не обнаружено увеличения содержания бластов в ПК и КМ. Так, у детей крайне редко диагностируются такие подтипы заболевания, как РАКС и МДС с изолированной del(5q) аномалией хромосом. Анемия, которая служит основным признаком, позволяющим заподозрить РА у взрослых, не так часто определяется у детей. В детском возрасте манифестация заболевания чаще связана с развитием нейтропении и тромбоцитопении [2]. У детей с МДС значительно чаще, чем у взрослых, наблюдается гипоклеточность КМ. Эти различия подчеркиваются введением в современную классификацию (ВОЗ, 2008) опухолей кроветворной
и лимфоидной тканей в качестве новой нозологической формы «Рефрактерная цитопения детского возраста (РЦДВ)».
РЦДВ (синоним — рефрактерная анемия детского возраста) — разновидность МДС, характеризующаяся персистентной цитопенией с наличием менее 5% бластов в КМ и менее 2% бластов в ПК [3]. Для установления диагноза РЦДВ требуется выявление диспластических изменений. Однако цитологическая оценка дисплазии — лишь один из аспектов установления морфологического диагноза. При диагностике РЦДВ обязательным является гистологическое изучение трепанобиоптата КМ. У 75% детей с РЦДВ наблюдается выраженная гипоклеточность КМ. При этом чрезвычайно важно дифференцировать РЦДВ и другие нарушения костномозгового кроветворения, особенно при приобретенной апластической анемии и наследственных нарушениях костномозгового кроветворения. РЦДВ — наиболее частый подтип МДС в детском возрасте. На его долю приходится почти 50% всех случаев заболевания. Диагностируется во всех возрастных группах, с одинаковой частотой у мальчиков и девочек. Предполагаемый источник возникновения — ГСК с мультилинейным потенциалом.
Наиболее частые клинические проявления — недомогание, кровоточивость, лихорадка, инфекции. Не отмечается наличия спленои гепатомегалии. Вторичная лимфаденопатия, связанная с локальным или системным инфекционным процессом. У 75% детей с РЦДВ содержание тромбоцитов в крови не превышает 150 х 10 9 /л, у 50% больных выявляется анемия. Как правило, наблюдается снижение количества лейкоцитов в ПК, а у 25% больных отмечается выраженная нейтропения. В мазках из пунктата КМ содержится менее 5% миелобластов, выявляются диспластические изменения в клетках двух различных линий миелопоэза или более чем в 10% клеток одной линии. В гистологических срезах трепанобиоптатов, являющихся нормоили гиперклеточными, отмечается некоторое усиление эритропоэза с увеличением количества незрелых клеток эритробластического ряда, в основном проэритробластов. Незрелые клетки эритробластического ряда образуют островки, содержащие не менее 10 клеток. Эти небольшие очаги эри-
тропоэза сочетаются с относительно равномерно рас
пределенными в срезах клетками гранулоцитарного ряда. Увеличивается количество мегалобластоидных клеток. Содержание бластов, не образующих кластеров, не достигает 5%, и для их идентификации используется иммуногистохимическое определение антигена CD34. Количество мегакариоцитов уменьшено, в них определяются диспластические изменения.
Для идентификации микромегакариоцитов в срезах трепанобиоптатов КМ при РЦДВ проводится иммуногистохимическое исследование с использованием моноклональных антител к антигену CD61 (гликопротеину IIIa), к антигену CD41 (гликопротеину IIb/IIIa) или фактору Виллебранда.
К числу наиболее частых цитогенетических аномалий относится моносомия 7. У большинства же больных РЦДВ, независимо от клеточного состава КМ, определяется нормальный кариотип. У больных с моносомией 7 отмечается более быстрое прогрессирование заболевания, чем у пациентов с трисомией 8 или нормальным кариотипом. В настоящее время у некоторых пациентов с РЦДВ эффективной является иммуносупрессивная терапия. Методом выбора может быть трансплантация ГСК.