Поликистоз почек
Поликистоз почек – заболевание характеризующееся образованием множественных кист (полостей, заполненные жидкостью) в тканях почек, которые приводят к нарушению их функции.

Поликистоз почек входит в группу кистозных заболеваний почек (КЗП).
В генетическом и клиническом отношении наследственные кистозные заболевания почек составляют гетерогенную группу. Они могут проявляться еще до рождения либо протекать бессимптомно в течение жизни. За последние годы отмечен существенный прогресс в изучении этиологии КЗП. Оказалось, что, несмотря на различную генетическую природу КЗП, процесс образования кист основан на схожих принципах.
КЛАССИФИКАЦИЯ КИСТОЗНЫХ ЗАБОЛЕВАНИЙ ПОЧЕК
К наследственным КЗП относятся:
АУТОСОМНО-ДОМИНАНТНЫЙ ПОЛИКИСТОЗ ПОЧЕК (АДПКП)
АДПКП является одним из часто встречающихся заболеваний человека (1 случай на 400 — 1000 людей, 12,5 млн. больных в мире). Около 5-10% взрослого населения, нуждающихся в заместительной почечной терапии (ЗПТ), страдают АДПКП.
КАК Я СЕБЯ БУДУ ЧУВСТВОВАТЬ ПРИ АДПКП?
Учитывая, что при данном типе заболевания прогрессирование очень медленное, вначале могут и не быть симптомов вообще. Часто первым признаком может стать высокое артериальное давление, кровь в моче, или чувство тяжести или боли в спине, боках, или брюшной полости. Иногда первым признаком является инфекция мочевыводящих путей и / или камни в почках.
Со временем, когда кисты будут расти, почки будут увеличиваться в размере и весе. В некоторых случаях, почки становятся настолько большими, что они вызывают увеличение объема живота.
ЧТО Я МОГУ СДЕЛАТЬ, ЧТОБЫ ЗАМЕДЛИТЬ ИЛИ ОСТАНОВИТЬ ПРОГРЕССИРОВАНИЕ АДПКП?
Есть несколько правил, при соблюдении которых, пациенты могут попытаться задержать наступление почечной недостаточности:
АУТОСОМНО-РЕЦЕССИВНЫЙ ПОЛИКИСТОЗ ПОЧЕК (АРПКП)
АРПКП встречается значительно реже, чем АДПКП. АРПКП относят к детским заболевания.
У новорожденных почки увеличены симметрично в размерах (иногда в 10 раз), сохраняя бобовидную форму.
В тяжелых случаях у плода развивается синдром Поттера, характеризующийся олигогидрамнионом (уменьшение околоплодных вод), легочной гипоплазией (недоразвитие легких), характерным хабитусом (внешностью), косолапостью. Из них до 50% умирают вскоре после рождения вследствие дыхательной и почечной недостаточности.
Одна треть детей с АРПКП, выжившых период новорожденности, может потребовать почечной заместительной терапии и почечной трансплантации до возраста 10 лет.
У 80% детей развивается артериальная гипертензия, коррекция которой может потребовать назначения препаратов (в частности препараты из группы ингибиторы Ангиотензин превращающего фермента (АПФ)). Тщательный мониторинг АД очень важен для предотвращения последствий гипертензии (гипертрофии сердца, застойной сердечной недостаточности и др.) и нарушения функции почек.
При УЗ-исследовании выявляется двустороннее увеличение почек с повышенной эхогенностью т плохой дифференциацией, наличие больших кист характерно для детей старшего возраста.
При рецессивном типе обязательным является наличие поражения печени, которое характеризуется врожденным фиброзом. В клинической картине заболевания, особенно у детей старшего возраста, могут превалировать гепатобилиарные осложнения (печеночные осложнения: портальная гипертензия, гиперспленизм (увеличение селезенки) с панцитопенией, развитие гнойного холангита, который может привести к фульминантной печеночной недостаточности).
Частота выявления мутаций у пациентов с полной клинической картиной АРПКП составляет 80%, а именно мутации гена PKHD1.
ПРЕНАТАЛЬНАЯ ДИАГНОСТИКА
В связи с возникновением заболевания в 25% случаев в семьях, где уже имеется больной ребенок, необходима ранняя пренатальная диагностика. Обычно по данным УЗИ заболевание обнаруживается в поздние сроки беременности или после рождения. Поэтому надежная пренатальная диагностика АРПКП в семьях из групп риска осуществима только с помощью молекулярно-генетического анализа гена PKHD1.
На сегодняшний день эффективной терапии АРПКП не существует, но разработаны методы, способные замедлить прогрессирование заболевания, что является первым существенным шагом.
Поликистоз почек на узи у плода что делать
В сроки 7-11 нед, по мере увеличения размеров плода, почки поднимаются на свое постоянное место в поясничной области за счет быстрого роста каудальной части эмбриона.
В начале почки состоят из нескольких, свободно распологающихся друг относительно друга долей, которые имеют тонкий корковый слой. В течение второго триместра доли сливаются, переставая быть разобщенными, корковый слой утолщается, но дольчатый контур почек продолжает сохраняться в течение нескольких лет после рождения.
Примерно в 11 нед гестации почки начинают выделять мочу, и с этого момента количество выделяемой мочи начинает постепенно увеличиваться.
Ко второму триместру беременности функция почек становятся основным фактором, влияющим на объем околоплодных вод. Их достаточный объем необходим для нормального развития легких и скелета плода, поскольку обеспечивает пространство для его роста и двигательной активности. Таким образом, функционирование мочевой системы является необходимым условием для нормального развития легких и костной системы плода.
Аномалии мочеполовой системы составляют около четверти всех врожденных пороков развития, при этом частота их распространенности при рождении составляет 0,2-0,6%. Аномалии мочеполового тракта возникают вследствие остановки развития на раннем этапе, аномалий развития почечных канальцев, нарушения процессов смещения почек в поясничную область, а также вследствие обструкции мочевыводящих путей или устья мочевого пузыря.
Чаще всего такие пороки имеют изолированный характер, но могут сочетаться с другими аномалиями развития плода или вызывать их. Сочетания мальформаций мочеполовой системы и других систем плода обнаруживается при широком спектре различных наследственных, в том числе связанных с хромосомными нарушениями, и спорадических синдромов. Кроме того, обструкции мочевыводящих путей, при которых уменьшается количество выделяемой мочи, приводят к маловодию и вследствие этого обусловливают деформации других частей тела плода. В частности, выраженное маловодие, которе возникает до 20 нед беременности, может вызвать гипоплазию легких, лицевые аномалии, в том числе приплюснутый нос, низкое расположение ушных раковин, косолапость и другие деформации конечностей.
Ультразвуковое исследование во втором и третьем триместрах беременности позволяет выявлять не менее 80-85% аномалий мочеполовой системы, что делает возможным проведение дородового консультирования будущих родителей и обеспечивает своевременное обследование и лечение сразу после родов. До появления эхографии именно запаздывание с лечением приводило к значительному нарушению функции почек или даже к ее полной потере.
Патологии почек у плода: что и когда показывает экспертное УЗИ при беременности
» data-image-caption=»» data-medium-file=»https://i2.wp.com/medcentr-diana-spb.ru/wp-content/uploads/2018/05/patologii-pochek-u-ploda.jpg?fit=450%2C300&ssl=1″ data-large-file=»https://i2.wp.com/medcentr-diana-spb.ru/wp-content/uploads/2018/05/patologii-pochek-u-ploda.jpg?fit=825%2C550&ssl=1″/>
Почечная система плода формируется из нервной трубки на 22-й день беременности и завершает закладку к 28 неделе. Не все женщины знают на ранних сроках о своём интересном положении и продолжают вести обычный образ жизни: занимаются спортом, ездят на отдых, переносят тяжести, принимают лекарства. В результате почечная система может пострадать, и патология не во всех случаях будет совместима с жизнью.
патологии почек у плода
» data-image-caption=»» data-medium-file=»https://i2.wp.com/medcentr-diana-spb.ru/wp-content/uploads/2018/05/patologii-pochek-u-ploda.jpg?fit=450%2C300&ssl=1″ data-large-file=»https://i2.wp.com/medcentr-diana-spb.ru/wp-content/uploads/2018/05/patologii-pochek-u-ploda.jpg?fit=825%2C550&ssl=1″ loading=»lazy» src=»https://i1.wp.com/medcentr-diana-spb.ru/wp-content/uploads/2018/05/patologii-pochek-u-ploda-825×550.jpg?resize=790%2C527″ alt=»патологии почек у плода» width=»790″ height=»527″ srcset=»https://i2.wp.com/medcentr-diana-spb.ru/wp-content/uploads/2018/05/patologii-pochek-u-ploda.jpg?resize=825%2C550&ssl=1 825w, https://i2.wp.com/medcentr-diana-spb.ru/wp-content/uploads/2018/05/patologii-pochek-u-ploda.jpg?resize=450%2C300&ssl=1 450w, https://i2.wp.com/medcentr-diana-spb.ru/wp-content/uploads/2018/05/patologii-pochek-u-ploda.jpg?resize=768%2C512&ssl=1 768w, https://i2.wp.com/medcentr-diana-spb.ru/wp-content/uploads/2018/05/patologii-pochek-u-ploda.jpg?w=900&ssl=1 900w» sizes=»(max-width: 790px) 100vw, 790px» data-recalc-dims=»1″/>
Причины аномального развития почек у плода
Гипоплазия или недоразвитость почки — это внутриутробная аномалия, при которой почка имеет маленькие размеры и неправильно функционирует. Отклонение встречается менее чем в 0,2% всех беременностей.
Развитию гипоплазии почки способствуют следующие факторы:
На УЗИ гипоплазия легко заметна, почки визуализируются с 14 недели беременности. В этом поможет высокоточный 3D аппарат. Почка отображается на экране монитора как овальное или бобовидное образование в продольном сканировании и округлое в поперечном.
Почка — это парный орган, расположенный по обе стороны от позвоночника. Основой органа является почечная лоханка, состоящая из сливающихся друг с другом почечных чашек. Лоханка плавно сужается и переходит в мочеточник, который ведёт в мочевой пузырь. Сам мочеточник у здорового плода не визуализируется на УЗИ.
У здорового органа диаметр почечной лоханки составляет 4-5 мм на 2 триместре и 7 мм на 3 триместре. Структурно-функциональной единицей почки является нефрон, который осуществляет фильтрацию. На 1 скрининге можно убедиться в наличии или отсутствии почек, в односторонней или двухсторонней недоразвитости (гипоплазии), удвоении почки, а также в нормальном или аномальном расположении. Но о функциональности органа станет ясно на 2 скрининге.
Какие патологии почек у плода можно выявить на 2 скрининге
2-й скрининг проводится на сроке 20-24 недели беременности. На нём выявляют различные пороки почки у плода:
Патология опасна тем, что на её фоне развивается почечная дисплазия — поражение почечной ткани кистами с нарушением функциональности органа. На УЗИ почка становится гиперэхогенной, в ней появляются кисты.
Выводы
Патология органов выделения составляет 1/4 часть всех пренатальных пороков развития. Ошибки возможны только в случае маловодия у женщины, когда органы плохо визуализируются.
Поликистозная болезнь почек с аутосомно-рецессивным типом наследования

Поликистозная болезнь почек с аутосомно-рецессивным типом наследования (ПБП-АР) принадлежит к группе врожденных гепаторенальных фибрознокистозных заболеваний. Первично всегда поражаются почки и печень, но с развитием заболевания в патологический процесс могут включаться и другие органы. Заболевание характеризуется двусторонним увеличением почек из-за наличия небольших множественных кист и поражением печени.
Эпидемиология
Младенческая форма. Встречается редко (1:10000–1:40000), частота может расти в изолированных популяциях. На данный момент не выявлено существующих гендерных и расовых предрасположенностей.
Этиология
Несмотря на клиническую вариабельность и множественное поражение органов, только единичная мутация гена PKHD1 в 6 хромосоме (6p12) отвечает за заболевание. Ген PKHD1 отвечает за экспрессию белка фиброцистина преимущественно в печени, почках и поджелудочной железе. Белок фиброцистин входит в состав первичных ресничек, располагающихся на апикальной мембране клеток эпителия почек и желчных путей.
Классификация
В зависимости от времени начала первых проявлений и степени поражения печени выделяют следующие типы заболевания:
1. Перинатальный тип — наиболее распространенный:
2. Неонатальный тип — минимальная степень перипортального фиброза печени;
3. Младенческий тип — умеренный перипортальный фиброз;
4. Ювенильный тип — выраженный перипортальный фиброз в сочетании с портальной гипертензией, спленомегалией и портосистемным варикозом.
Патологическая анатомия
Макроскопическое строение
Плод имеет характерный внешний вид (лицо Поттера): узкие щели век, характерная борозда под линией века, микрогнатия, приплюснутый нос, мягкие большие уши аномальной формы. Почки увеличены в размерах, при этом сохраняют свою бобовидную форму, но не могут выполнять свою функцию. Плохо функционирующие почки не производят достаточное количество фетальной мочи, что приводит к маловодию и гипоплазии легких. Гидростатическое давление амниотической жидкости обеспечивает нормальное развитие дыхательной системы.
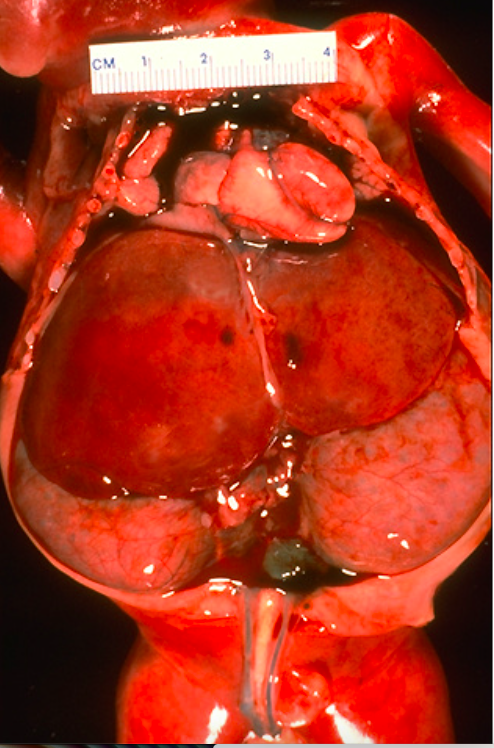
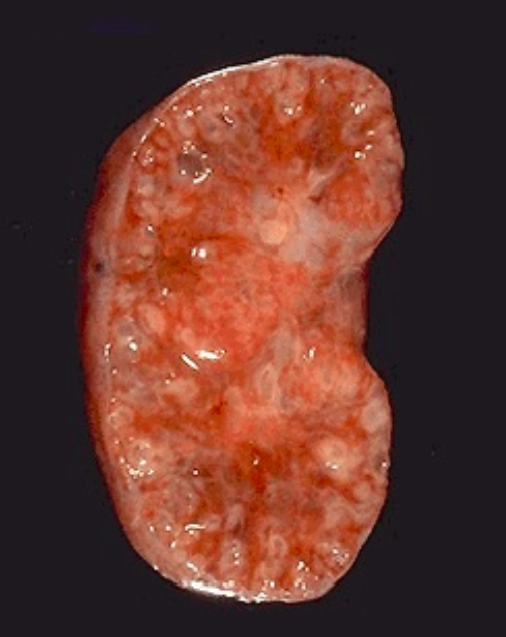
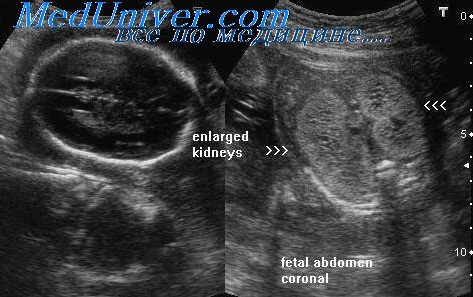
Рисунок 2 | Продольный срез почки с ПКБ.
Микроскопия
Почки
Радиально ориентированные расширенные собирательные канальцы формируют почечные кисты диаметром 1–2 мм, между которыми видны нормальные клубочки и канальцы. Размеры кист могут варьировать в зависимости от возраста. На ранних стадиях болезнь дебютирует с микрокист, которые затем растут и превращаются в макрокисты. Кистозное поражение почек сопровождается незначительным интерстициальным фиброзом паренхимы почек.
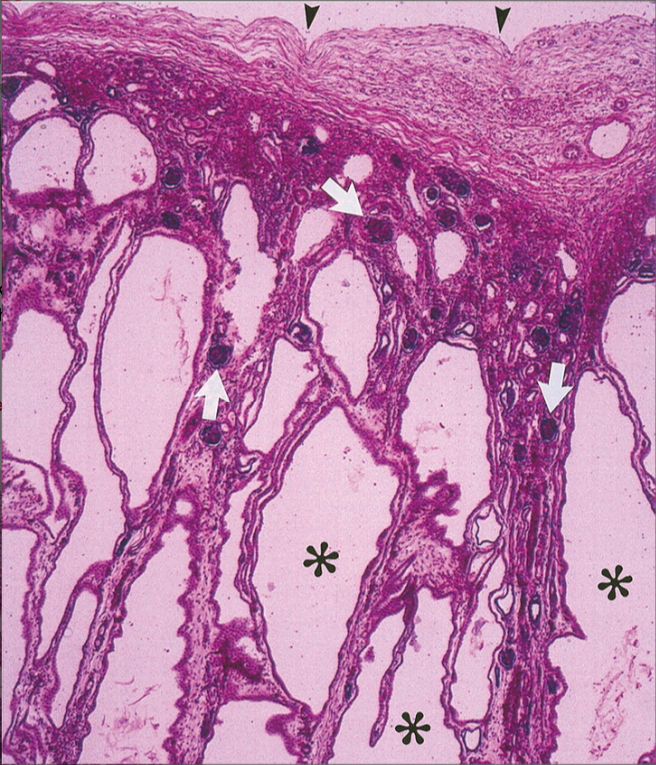
Рисунок 3 | Микропрепарат почки пациента с ПБК-АР. Двадцатикратное увеличение, окраска гематоксилин-эозин.
✱ — радиальные почечные кисты;
▼— почечная капсула.
Стрелками обозначены нормальные клубочки между расширенными собирательными канальцами.
Печень
Гистологические изменения печени включают следующие пороки развития дуктальной пластинки: гиперплазию желчевыводящих путей, билиарную эктазию и перипортальный фиброз. Нарушения морфогенеза развития желчных путей приводит к их дилатации. С последующим прогрессированием заболевания расширенные протоки превращаются в макрокисты, связанные нормальными протоками, что позволяет достаточно хорошо их верифицировать с помощью МРХПГ.
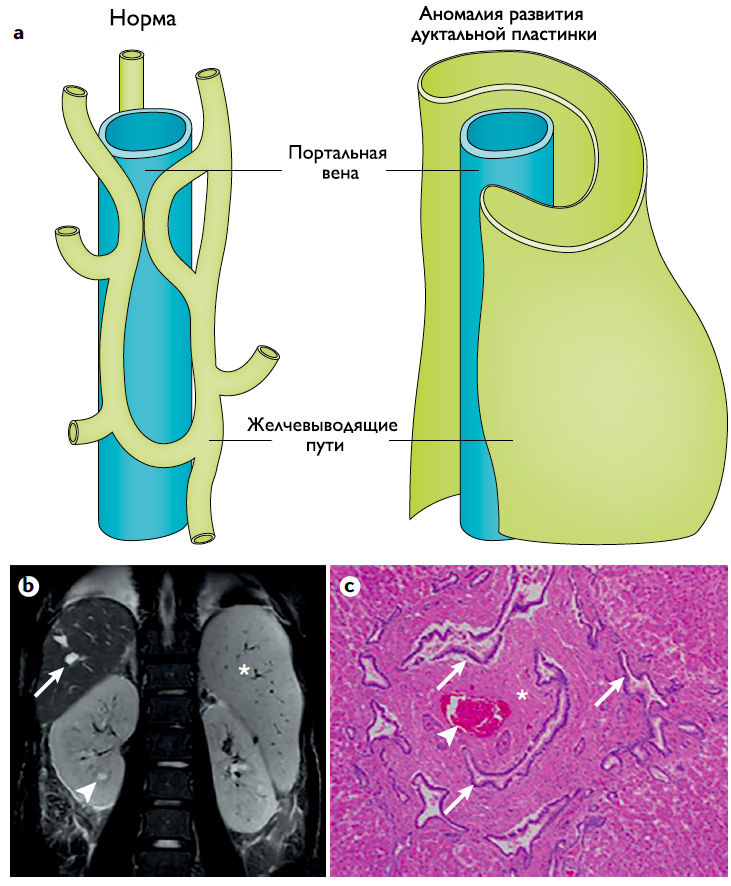
Рисунок 4 | Патологические изменения печени.
a) Строение нормально разветвленной портовенозной и решетчатой систем желчных путей (слева) нарушается вследствие дефекта развития дуктальной пластинки и ошибок в терминальной дифференцировке холангиоцитов (справа);
b) Корональное Т2-взвешенное изображение брюшной полости:
— стрелкой показано кистозное веретенообразное расширение желчных протоков;
— стрелкой по типу наконечника стрелы — нефромегалия с маленькими цистами;
✱ — спленомегалия.
c) Препарат фрагмента печени, окраска гематоксилин-эозин, увеличение в 40 раз:
Формирование кист
При ПБП-АР кисты формируются преимущественно из дистальных канальцев и собирательных трубочек, детальные молекулярные механизмы этого до сих остаются покрыты мраком. В результате неправильного восприятия сигналов из-за нарушенной работы первичных ресничек в клетках почечного эпителия происходит нарушение внутренних сигнальных механизмов пролиферации и дифференцировки, что приводит к образованию пузырьков, наполненных жидкостью.
Клиническая характеристика
Первые симптомы у большинства пациентов проявляются в неонатальном периоде.
Почки
При осмотре: увеличение правых и левых боковых областей живота, где пальпируются объемные образования. Объем выделяемой мочи обычно уменьшен, хотя при нарушении концентрационной функции почек могут иметь место явления полиурии и полидипсии. Тяжелые формы с развитием острой почечной недостаточности (ОПН) будут проявляться олигурией, гипонатриемией, увеличением креатинина и азота мочевины в крови. Но с возрастом благодаря развитию нормальной почечной ткани происходит улучшение почечной функции и уменьшение проявлений ОПН. Несмотря на это, примерно у 50 % пораженных в первую декаду жизни болезнь прогрессирует в терминальную почечную недостаточность. Симптомы тяжелой артериальной гипертензии, которая дебютирует с первых дней, также становятся менее тяжелыми с течением времени.
Печень
С развитием методов заместительной терапии функций почек и трансплантологии улучшается долгосрочная выживаемость пациентов. Благодаря этому гепатобилиарные клинические проявления становятся основной проблемой старших возрастных групп. Врожденный фиброз печени приводит к прогрессирующей портальной гипертензии, что проявляется варикозно расширенными венами пищевода, желудка, геморроидальных вен, гепатоспленомегалией, энтеропатией с потерей белка и желудочно-кишечными кровотечениями.
Синдром Кароли— необструктивная дилатация внутрипеченочных желчных протоков и общего желчного протока, встречающаяся более чем в 60 % случаев ПКБ-АР. Недостаточность дренажной функции желчной системы предрасполагает к развитию бактериального восходящего холангита и сепсиса. Мальабсорбция из-за холестаза приводит к дефициту жирорастворимых витаминов (A, D, E и K). У взрослых пациентов гиперплазия желчевыводящих путей предрасполагает к возникновению добро- и злокачественных опухолей.
Диагностика
Диагноз ПБП-АР должен быть заподозрен во всех случаях диффузного эхогенного увеличения обеих почек при УЗИ. Для постановки точного диагноза обычно бывает достаточно клинических признаков и наличия радиографических изменений. Но для точной верификации заболевания разработаны специальные диагностические критерии: сочетание патогномоничных признаков изменения почки — одного или нескольких пунктов из следующих:
Ультразвуковое исследование
УЗИ высокого разрешения значительно улучшило диагностику легких форм заболевания, когда клинические проявления невыраженные, обеспечивая неинвазивную детальную визуализацию изменений в почках без использования радиации и контрастных веществ.
Ультрасонография является методом выбора при исследовании плода и детских форм ПКБ-АР. УЗИ диагностика обладает большим перечнем преимуществ: низкая стоимость, безболезненность, широкая распространенность и отсутствие необходимости в излучении и седации. Но несмотря на все перечисленные достоинства УЗИ, с целью верификации диагностика дополняется МРТ.
Патогномоничные изменения почек, диагностируемые при помощи УЗИ:
Младенчество (до года):
Детство и юношеский возраст:
Рисунок 5 | Обе почки увеличены в продольном размере (правая 10 см и левая 9 см), с повышенной эхогенностью (за счет акустического усиления крошечных кист) и измененной кортикомедуллярной дифференцировкой.
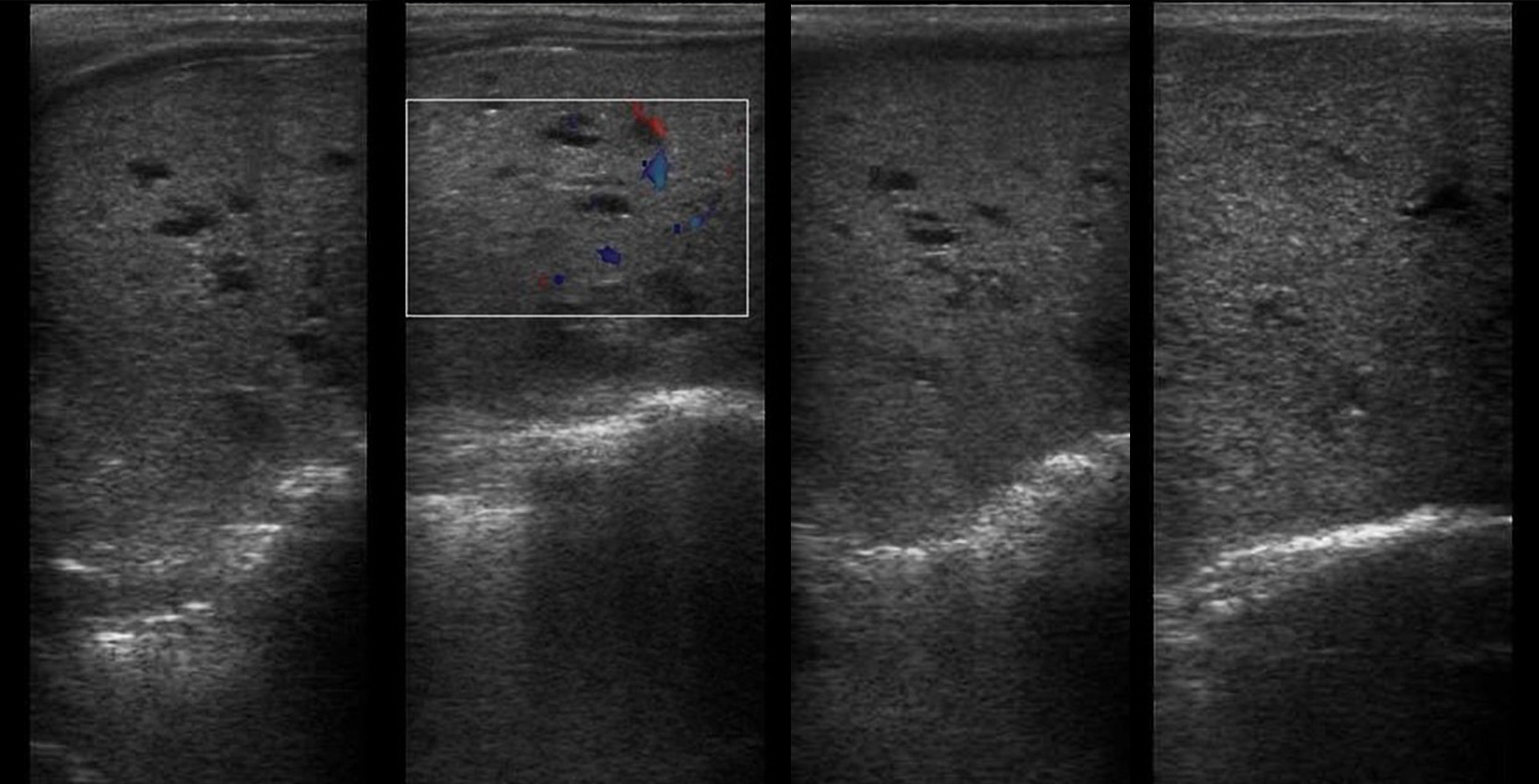
Рисунок 6 | Печень с врожденным фиброзом и кистозными расширениями в правой доле.
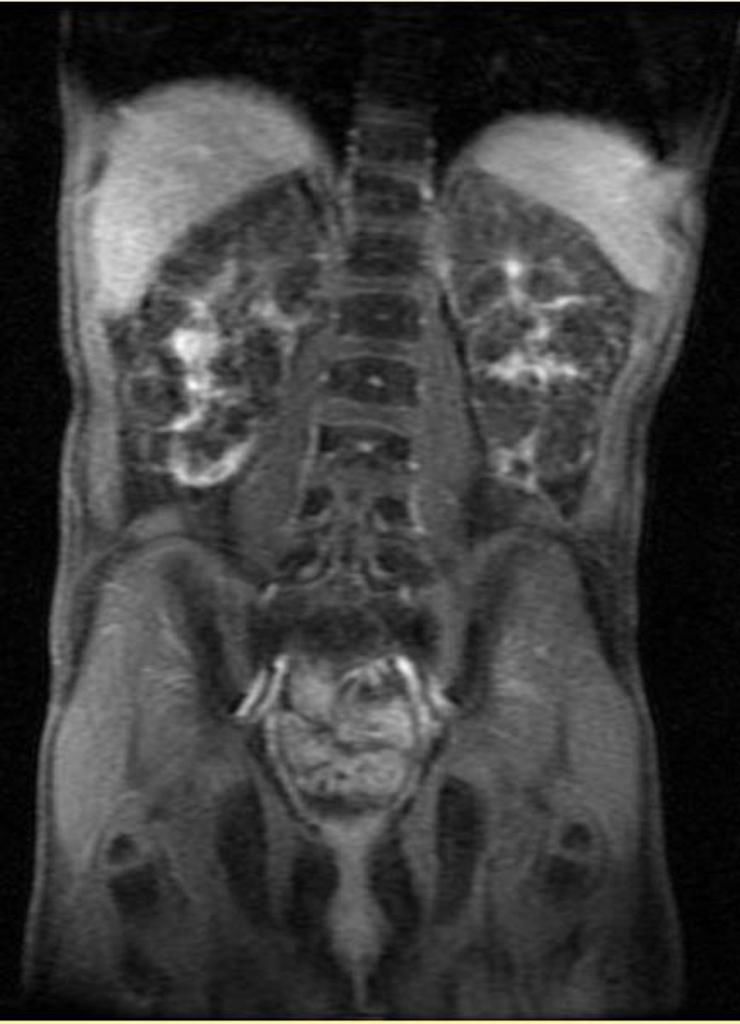
Рисунок 7 | МРТ, корональная Т2-проекция, пациент с ПБП-АР.
Магнитно-резонансный томограф не имеет никаких преимуществ перед ультразвуковыми сканерами высокого класса и генетическим тестированием в диагностике ПКП-АД.
Магнитно-резонансная холангиопанкреатография (МРХПГ)
Изображения, получаемые с помощью МРХПГ, обеспечивают хорошее качество визуализации гепатобилиарной системы у пациентов с ПБП-АР. МРХПГ является чувствительным методом диагностики билиарной эктазии, который в сочетании с визуализацией почек почти полностью заменил такие инвазивные методы диагностики, как биопсия почек и печени.
NB! Биопсия почек и печени не должны использоваться для диагностики ПБП.
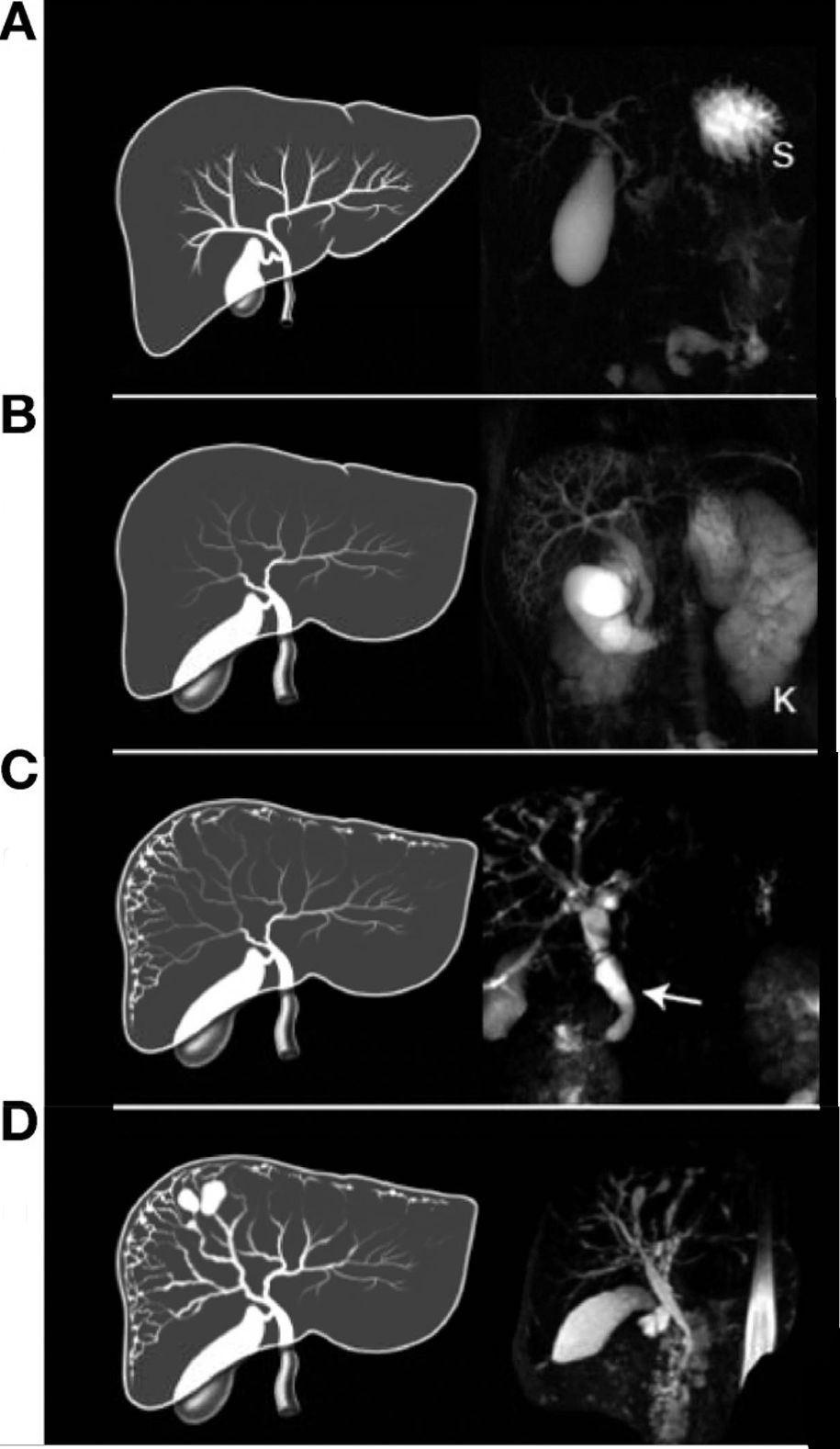
Рисунок 8 | МРХПГ и последующий рендеринг билиарной системы с аномалиями, свойственными для пациентов с ПБП-АР:
A. Нормальная печень с увеличенным желчным пузырем, (S) желудок.
B. Увеличенный общий желчный проток с желчным пузырем. Расширенные внутрипеченочные протоки.
C. Веретенообразные кисты периферических и центральных желчных протоков небольшого размера с расширенным общим желчным протоком (стрелка) и желчным пузырем.
D. Веретенообразные макроцисты с расширенными печеночными протоками.
У всех пациентов была печень аномальной формы с непропорционально увеличенной левой долей.
Дифференциальная диагностика
ПБП-АР следует дифференцировать со следующими состояниями:
Подтверждение диагноза
Если имеются серьезные подозрения на ПБП-АР, необходим генетический анализ и консультирование. Молекулярно-генетическое тестирование показано пробанду с наличием у него кистозного увеличения почек и ВФП для выявления обеих патологических аллелей PKHD1. Должны быть проанализированы как минимум 3 поколения, т. к. заболевание передается рецессивно. Молекулярно-генетическое подходы к тестированию включают в себя панели для диагностики единичных точечных мутаций, мультигенные панели или полное геномное исследование.
Лечение
Аппарат искусственного кровообращения необходим всем пациентам с гипоплазией легкого, у которых оксигенация 100 % кислородом неэффективна из-за гипервентиляции вследствие давления на диафрагму. Новорожденным с врожденной легочной недостаточностью может потребоваться ИВЛ с первых минут жизни до определения причины. Когда увеличенные почки сдавливают ЖКТ, ряд авторов рекомендуют двух- или одностороннюю нефрэктомию, но при односторонней нефрэктомии может наблюдаться компенсаторное еще большее увеличение оставшейся почки. После двусторонней нефрэктомии пациенты нуждаются в гемодиализе.
Хронический перитонеальный диализ, с короткими периодами на гемодиализ для восстановления брюшины — метод выбора. Продолжительность этих процедур, также как и превентивная трансплантация почки, будут зависеть от ряда таких факторов как возраст, вес, клинический статус пациента и наличие здорового подходящего донора. Лечение гипонатриемии должно проводиться в зависимости от степени обезвоживания — раннее распознавание и лечение дегидратации имеют решающее значение. Для введения дополнительного питания и жидкостей может потребоваться установка назогастрального зонда. Артериальная гипертония (АГ) обычно хорошо поддается лечению ингибиторами АПФ и блокаторами рецепторов ангиотензина, которые являются препаратами выбора. Порой АГ может потребовать сочетания нескольких препаратов. Для лечения анемии у детей с хронической почечной недостаточностью необходимо добавление препаратов железа или стимуляторов выработки эритропоэтина. Лечение билиарной дисфункции должно быть сфокусировано на мальабсорбции питательных веществ и жирорастворимых витаминов и снижении риска восходящего холангита. Варикозно расширенные вены пищевода можно клипировать или использовать склеротерапию. Прогрессирующая портальная гипертензия требует портосистемного шунтирования. Благодаря возможностям современной медицины, в тяжелых случаях портальной гипертензии, почечной и печеночной недостаточности стала возможна двойная почечно-печеночная трансплантация.
Поликистоз почек у детей
Поликистоз почек у детей встречается с частотой 1 случай на 250-1000 новорожденных, однако в связи с латентным течением в детском возрасте диагностируется редко. В зависимости от клинической формы, поликистоз почек может обнаружиться или проявиться в различные возрастные периоды: у новорожденных, детей раннего возраста, подростков или взрослых преимущественно в возрасте 30-40 лет. Кистозные изменения почек у детей нередко сочетаются с поликистозом печени, поджелудочной железы, селезенки, легких, поликистозными яичниками, мегауретером, добавочной почкой.
Причины и симптомы
Детскийполикистоз почек формируется уже в самом начале эмбрионального развития и обусловливается несрастанием канальцев метанефроса (окончательной почки) и собирательных канальцев зачатка мочеточника. Наряду с наследственным фактором, возникновение поликистоза связывают и с другими причинами. Мутации генов могут быть связаны с воздействием лекарств и химических веществ, разнообразных вирусов и иных отрицательных факторов.
В зависимости от того, в каком возрасте у ребенка появились первые симптомы заболевания, выделяют следующие группы:
Перинатальную (начинается с 28 недели беременности и заканчивается через 7 дней после родов).
Неонатальную (первые 4 недели жизни после рождения).
Раннего детского возраста (3-6 мес.).
Ювенильную (6 мес.5 лет).
При поликистозе 3-4 групп количество кист намного меньше, но возникают признаки аномалии других органов.
Самыми частыми симптомами детского поликистоза являются: артериальная гипертензия (повышение артериального давления), инфекции мочевых путей. Кроме того, со временем развивается почечная недостаточность, которая проявляется в виде нарушений работоспособности почек, анемии, изменений в костной ткани, отставании малыша в росте. Вместе с этим обнаруживается печеночный фиброз (разрастание соединительной ткани), часто осложненный кровотечениями в пищеводе и желудочно-кишечном тракте, а также повышением давления в системе воротной вены.
Диагностика
Диагноз ставится посредством проведения инструментального и клинико-лабораторного обследования малыша, а также анализа его родословной. Необходимо проведение общего анализа мочи, исследования мочи по методике Зимницкого, пробы Реберга, биохимии крови.
С целью подтверждения диагноза проводится экскреторная урография (рентгенологический метод исследования мочевыводящих путей), почечная ангиография (метод контрастного рентгенологического исследования кровеносных сосудов), динамическая сцинтиграфия (метод функциональной визуализации для получения двумерного изображения), УЗИ, компьютерная томография.
С целью выявления мутантного гена применяется метод молекулярной гибридизации (объединение генетического материала разных клеток в одной клетке). В настоящее время недуг можно выявить внутриутробно на УЗИ (после 30-ой недели).
Лечение
Лечение детскогополикистоза осуществляется на протяжении всей жизни. Оно направлено на предупреждение осложнений и улучшение работы почек. Больные детки нуждаются в дополнительных курсах терапии. Кроме того, они должны постоянно соблюдать питьевую и пищевую диеты. В случае сопутствующего воспаления в почках осуществляется терапия пиелонефрита с помощью уросептиков и препаратов группы антибиотиков. Если присоединяется артериальная гипертензия, прописываются гипотензивные препараты.При развитии хронической формы почечной недостаточности на фоне поликистоза осуществляется гемодиализ (очищение крови) и решается вопрос о процедуре трансплантации почки.
При увеличении объема кист может понадобиться оперативное вмешательство: чрескожная пункционная аспирация (введение иглы) кист, а также лапараскопическое иссечение кистозных образований.