Амилоидоз
МКБ-10
Общие сведения
Причины амилоидоза
Патогенез
Среди многочисленных версий амилоидогенеза наибольшее число сторонников имеют теория диспротеиноза, локального клеточного генеза, иммунологическая и мутационная теории. Теория локального клеточного генеза рассматривает лишь процессы, происходящие на клеточном уровне (образование фибриллярных предшественников амилоида системой макрофагов), в то время как образование и накопление амилоида происходит вне клетки. Поэтому теория локального клеточного генеза не может считаться исчерпывающей.
Амилоид представляет собой сложный гликопротеид, состоящий из фибриллярных и глобулярных белков, тесно связанных с полисахаридами. Амилоидные отложения накапливаются в интиме и адвентиции кровеносных сосудов, строме паренхиматозных органов, железистых структурах и т. д. При незначительных отложениях амилоида изменения выявляются лишь на микроскопическом уровне и не приводят к функциональным нарушениям. Выраженное скопление амилоида сопровождается макроскопическими изменениями пораженного органа (увеличением объема, сальным или восковым видом). В исходе амилоидоза развивается склероз стромы и атрофия паренхимы органов, их клинически значимая функциональная недостаточность.
Классификация
В соответствии с причинами различают первичный (идиопатический), вторичный (реактивный, приобретенный), наследственный (семейный, генетический) и старческий амилоидоз. Встречается различные формы наследственного амилоидоза: средиземноморская лихорадка, или периодическая болезнь (приступы жара, боли в животе, запор, диарея, плеврит, артрит, высыпания на коже), португальский нейропатический амилоидоз (периферическая полинейропатия, импотенция, нарушения сердечной проводимости), финский тип (атрофия роговицы, краниальная невропатия), датский вариант (кардиопатический амилоидоз) и мн. др.
В зависимости от преимущественного поражения органов и систем выделяют нефропатический (амилоидоз почек), кардиопатический (амилоидоз сердца), нейропатический (амилоидоз нервной системы), гепатопатический (амилоидоз печени), эпинефропатический (амилоидоз надпочечников), АРUD-амилоидоз, амилоидоз кожи и смешанный тип заболевания. Кроме этого, в международной практике принято различать локальный и генерализованный (системный) амилоидоз. К локализованным формам, как правило, развивающимся у лиц старческого возраста, относятся амилоидоз при болезни Альцгеймера, сахарном диабете 2-го типа, эндокринных опухолях, опухолях кожи, мочевого пузыря и др. В зависимости от биохимического состава амилоидных фибрилл среди системных форм амилоидоза выделяют следующие типы:
Симптомы амилоидоза
Клинические проявления амилоидоза отличаются многообразием и зависят от выраженности и локализации амилоидных отложений, биохимического состава амилоида, «стажа» заболевания, степени нарушения функции органов. В латентной стадии амилоидоза, когда отложения амилоида могут быть обнаружены только микроскопически, симптоматика отсутствует. По мере развития и прогрессирования функциональной недостаточности того или иного органа нарастают клинические признаки заболевания.
При амилоидозе почек длительно текущая стадия умеренной протеинурии сменяется развитием нефротического синдрома. Переход к развернутой стадии может быть связан с перенесенной интеркуррентной инфекцией, вакцинацией, переохлаждением, обострением основного заболевания. Постепенно нарастают отеки (сначала на ногах, а затем на всем теле), развивается нефрогенная артериальная гипертензия и почечная недостаточность. Возможно возникновение тромбоза почечных вен. Массивная потеря белка сопровождается гипопротеинемией, гиперфибриногенемией, гиперлипидемией, азотемией. В моче обнаруживается микро-, иногда макрогематурия, лейкоцитурия. В целом в течение амилоидоза почек выделяют раннюю безотечную стадию, отечную стадию, уремическую (кахектическую) стадию.
Амилоидоз сердца протекает по типу рестриктивной кардиомиопатии с типичными клиническими признаками – кардиомегалией, аритмией, прогрессирующей сердечной недостаточностью. Больные жалуются на одышку, отеки, слабость, возникающую при незначительных физических нагрузках. Реже при амилоидозе сердца развивается полисерозит (асцит, экссудативный плеврит и перикардит).
Поражение ЖКТ при амилоидозе характеризуется амилоидной инфильтрацией языка (макроглассией), пищевода (ригидностью и нарушением перистальтики), желудка (изжогой, тошнотой), кишечника (запорами, диареей, синдромом мальабсорбции, кишечной непроходимостью). Возможно возникновение желудочно-кишечных кровотечений на различных уровнях. При амилоидной инфильтрации печени развивается гепатомегалия, холестаз, портальная гипертензия. Поражение поджелудочной железы при амилоидозе обычно маскируется под хронический панкреатит.
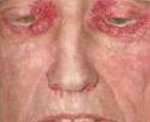
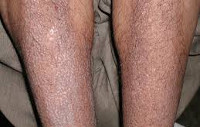
Амилоидоз кожи протекает с появлением множественных восковидных бляшек (папул, узелков) в области лица, шеи, естественных кожных складок. По внешним признакам поражение кожи может напоминать склеродермию, нейродермит или красный плоский лишай. Для амилоидного поражения опорно-двигательного аппарата типично развитие симметричного полиартрита, запястного туннельного синдрома, плечелопаточного периартрита, миопатии. Отдельные формы амилоидоза, протекающие с вовлечением нервной системы, могут сопровождаться полинейропатией, параличами нижних конечностей, головными болями, головокружением, ортостатической гипотензией, потливостью, деменцией и т. д.
Диагностика
С клиническими проявлениями амилоидоза могут столкнуться различные клиницисты: ревматологи, урологи, кардиологи, гастроэнтерологи, неврологи, дерматологи, терапевты и др. Первостепенное значение для правильной постановки диагноза имеет всесторонняя оценка клинических и анамнестических признаков, проведение комплексного лабораторного и инструментального обследования.
С целью оценки функционального состояния сердечно-сосудистой системы назначается ЭхоКГ, ЭКГ. Обследование органов пищеварительного тракта предполагает проведение УЗИ органов брюшной полости, рентгеновской диагностики (рентгенография пищевода, желудка, ирригография, рентгенография пассажа бария), эндоскопических исследований (ЭГДС, ректороманоскопия). Об амилоидозе следует думать при сочетании протеинурии, лейкоцитурии, цилиндрурии с гипопротеинемией, гиперлипидемией (повышением в крови содержания холестерина, липопротеидов, триглицеридов), гипонатриемией и гипокальциемией, анемией, снижением количества тромбоцитов. Электрофорез сыворотки крови и мочи позволяет определить наличие парапротеинов.
Окончательная диагностика амилоидоза возможна после обнаружения амилоидных фибрилл в пораженных тканях. С этой целью может производиться биопсия почки, лимфатических узлов, десен, слизистой оболочки желудка, прямой кишки. Установлению наследственного характера амилоидоза способствует тщательный медико-генетический анализ родословной.
Лечение амилоидоза
Отсутствие полноты знаний об этиологии и патогенезе заболевания обусловливают трудности, связанные с лечением амилоидоза. При вторичном амилоидозе важное значение имеет активная терапия фонового заболевания. Рекомендации по питанию предполагают ограничение приема поваренной соли и белка, включение в рацион сырой печени. Симптоматическая терапия амилоидоза зависит от наличия и выраженности тех или иных клинических проявлений. В качестве патогенетической терапии могут назначаться препараты 4-аминохинолинового ряда (хлорохин), диметилсульфоксид, унитиол, колхицин. Для терапии первичного амилоидоза используются схемы лечения цитостатиками и гормонами (мельфолан+преднизолон, винкристин+доксорубицин+дексаметазон). При развитии ХПН показан гемодиализ или перитонеальный диализ. В отдельных случаях ставится вопрос о трансплантации почек или печени.
Прогноз
Течение амилоидоза носит прогрессирующий, практически необратимый характер. Заболевание может отягощаться амилоидными язвами пищевода и желудка, кровотечениями, печеночной недостаточностью, сахарным диабетом и др. При развитии хронической почечной недостаточности средняя продолжительность жизни больных составляет около 1 года; при развитии сердечной недостаточности – около 4 месяцев. Прогноз вторичного амилоидоза определяется возможностью терапии основного заболевания. Более тяжелое течение амилоидоза отмечается у пожилых пациентов.
Клинические рекомендации по диагностике и лечению системного амилоидоза
В клинических рекомендациях, подготовленных специалистами различного профиля, рассматриваются методы диагностики и лечениясистемного амилоидоза, в том числе АА (вто-ричный амилоидоз при хронических воспалительных заболеваниях, включая ревматоидныйартрит, анкилозирующий спондилит, аутовоспалительные заболевания, хроническиенагноения, злокачественные опухоли и др.), AL (амилоидоз при плазмоклеточных дискразиях – идиопатический, при миеломной болезни и макроглобулинемии Вальденстрема) иATTR (транстиретиновый; семейные формыполиневропатического, кардиопатического идругого амилоидоза, системный старческийамилоидоз). Диагноз амилоидоза, которыйможно заподозрить на основании клиническихданных, необходимо подтвердить при гистологическом исследовании (окрашивание препаратов ткани конго-красным с последующей микроскопией в поляризованном свете). Чтобы замедлить или приостановить прогрессирование амилоидоза любого типа, необходимо добиться уменьшения количества (или, если возможно, удаления) белков-предшественников путем лечения хронического воспаленияпри АА-амилоидозе или подавления пролиферации клона плазматических клеток для уменьшения продукции легких цепей иммуно-глобулинов при AL-амилоидозе. Для замедления прогресирования ATTR-амилоидоза упациентов с полиневропатией применяют тафамидис, который ингибирует диссоциацию мутантного транстиретина и снижает его амилоидогенность.
Определение, классификация, группы риска и принципы диагностики
Амилоидоз – группа заболеваний, отличительным признаком которых является отложение в тканях и органах фибриллярного гликопротеида амилоида. Специфическое свойство амилоида, отличающее его от других фибриллярных белков стромы, – способность к двойному лучепреломлению, что проявляется свечением в поляризованном свете предварительно окрашенных конгокрасным препаратов амилоида с изменением красного цвета конгофильных амилоидных отложений на яблочно-зеленый (дихроизм).
В основе амилоидогенеза лежит синтез большого количества нестабильных белковпредшественников, которые агрегируются с образованием амилоидной фибриллы. Клю чевое значение имеет амилоидогенность основного белка-предшественника амилоида, специфичного для каждой формы амилоидоза (в настоящее время известно более 30 таких белков), обозначение которого положено в основу современной классификации заболевания (ВОЗ, 2016 г.). Названия типов амилоида включают в себя букву А, означающую “амилоид», и обозначение конкретного фибриллярного белка амилоида – А (амилоидный А-протеин), L (легкие цепи иммуноглобулинов), TTR (транстиретин), β2М (β2-микроглобулин), В (В-протеин), IAPP (островковый амилоидный полипептид). Используют также производные наименования – иммуноглобулиновый амилоидоз (AL), транстиретиновый (ATTR) и др. (табл. 1) 3. Следует отметить, что Международная классификация болезней (МКБ) 10-го пересмотра базируется на клиническом принципе, не учитывает особенности патогенеза различных форм амилоидоза и не позволяет обосновать адекватное лечение.
| Белок амилоида | Белок-Белок-предшественник | Клиническая форма амилоидоза |
|---|---|---|
| АА | SSA-белок | Вторичный амилоидоз при хронических воспалительных заболеваниях, в том числе периодической болезни и синдроме Макла-Уэллса |
| AL | λ, κ-легкие цепи иммуноглобулинов | Амилоидоз при плазмоклеточных дискразиях – идиопатический, при миеломной болезни и макроглобулинемии Вальденстрема |
| ATTR | Транстиретин | Семейные формы полиневропатического, кардиопатического и др. амилоидоза, системный старческий амилоидоз |
| Аβ2М | β2-микроглобулин | Диализный амилоидоз |
| AGel | Гелсолин | Финская семейная амилоидная полиневропатия |
| AApoAI | Аполипопротеин А-I | Амилоидная полиневропатия (III тип по van Allen, 1956 г.) |
| AFib | Фибриноген | Амилоидная нефропатия |
| Aβ2 | β-белок | Болезнь Альцгеймера, синдром Дауна, наследственные кровоизлияния в мозг с амилоидозом |
| APrPScr | Прионовый белок | Болезнь Крейтцфельда-Якоба, болезнь Герстманна-Штраусслера-Шейнкера |
| AANF | Предсердный натрийуретический фактор | Изолированный амилоидоз предсердий |
| AIAPP | Амилин | Изолированный амилоидоз в островках Лангерганса при сахарном диабете 2 типа, инсулиноме |
| ACal | Прокальцитонин | При медуллярном раке щитовидной железы |
| ACys | Цистатин С | Наследственные кровоизлияния в мозг с амилоидозом (Исландия) |
АА-амилоидоз чаще всего развивается при ревматоидном артрите, серонегативных спондилоартропатиях, аутовоспалительных наследственных периодических лихорадках, в том числе периодической болезни (семейной средиземноморской лихорадке), а также при хронических нагноениях, туберкулезе. АА-амилоид образуется из сывороточного предшественника SAA (serum amyloid A) – острофазового белка, продуцируемого в значительных количествах в ответ на воспаление. По этой причине АА-амилоидоз называют также реактивным или вторичным.
Клинические формы AL-амилоидоза обусловлены единым этиологическим фактором – В-лимфоцитарной дискразией, характеризующейся формированием аномального клона плазматических или В-клеток в костном мозге, которые продуцируют аномальные иммуноглобулины, обладающие амилоидогенностью (легкие цепи моноклонального иммуноглобулина, чаще λ, реже κ-типа). При первичном AL-амилоидозе плазмоклеточная дискразия относительно более доброкачественная, в то время как при В-гемобластозах (множественной миеломе, болезни Вальденстрема и др.) она обладает признаками злокачественной опухоли. Аномальный амилоидогенный клон плазматических клеток может формироваться также из плазмоцитов, локализующихся вне костного мозга, что может привести к развитию локального амилоидоза. Наиболее распространенные локальные формы AL-амилоидоза – амилоидоз трахеи, бронхов и гортани, мочевого пузыря. Выявление плазмоклеточной дискразии необходимо для диагностики AL-амилоидоза, а также для оценки его риска и дифференциального диагноза.
ATTR-амилоидоз является необратимо прогрессирующим заболеванием с высокой степенью инвалидизации вследствие тяжелого поражения сердца, периферической и/или автономной полиневропатии. Пациенты обычно умирают в течение 10-12 лет от первых проявлений. Развитие ATTR-амилоидоза обусловлено мутациями в молекуле транстиретина или возрастным нарушением секреции тетрамеров транстиретина печенью. В обоих случаях происходит распад тетрамеров транстиретина до мономеров, обладающих выраженной конформационной нестабильностью.
Рекомендации:
Клинические проявления
Для вторичного АА-амилоидоза характерно более раннее начало, чем для AL-амилоидоза (средний возраст больных составляет около 40 и 65 лет, соответственно). ATTR-амилоидоз, несмотря на наследственную природу, характеризуется низкой пенетрантностью и также проявляется обычно после 35 лет.
Поражение почек – ведущий клинический признак АА- и AL-амилоидоза, наблюдающийся практически у всех больных. Поражение почек встречается и у больных с многими формами семейного амилоидоза (AFib, ALys, AGel и др.). При ATTR-амилоидозе нефропатия отмечается лишь у 20-23% больных. Клинически амилоидная нефропатия характеризуется неуклонно прогрессирующим течением с последовательной сменой стадий: протеинурия, нефротический синдром, хроническая почечная недостаточность (ХПН). Иногда возможно развитие ХПН без предшествующего нефротического синдрома.
Поражение сердца развивается у подавляющего большинства больных AL-амилоидозом и у 50-60% пациентов с АTTR-амилоидозом, но не характерно для АА-амилоидоза (рис. 1). При эхокардиографии у больных амилоидозом сердца наблюдается утолщение межжелудочковой перегородки и стенки левого желудочка (чаще симметричное), которое не сопровождается электрокардиографическими признаками гипертрофии миокарда. У части больных отмечается снижение вольтажа зубцов на ЭКГ, хотя отсутствие этого признака не исключает диагноз амилоидоза сердца. Нарушение диастолической функции левого желудочка (рестриктивный тип) приводит к развитию сердечной недостаточности, которая быстро прогрессирует, плохо поддается лечению и почти у 50% пациентов оказывается причиной смерти. Кроме того, у больных амилоидозом сердца часто наблюдаются различные аритмии и нарушения проводимости.
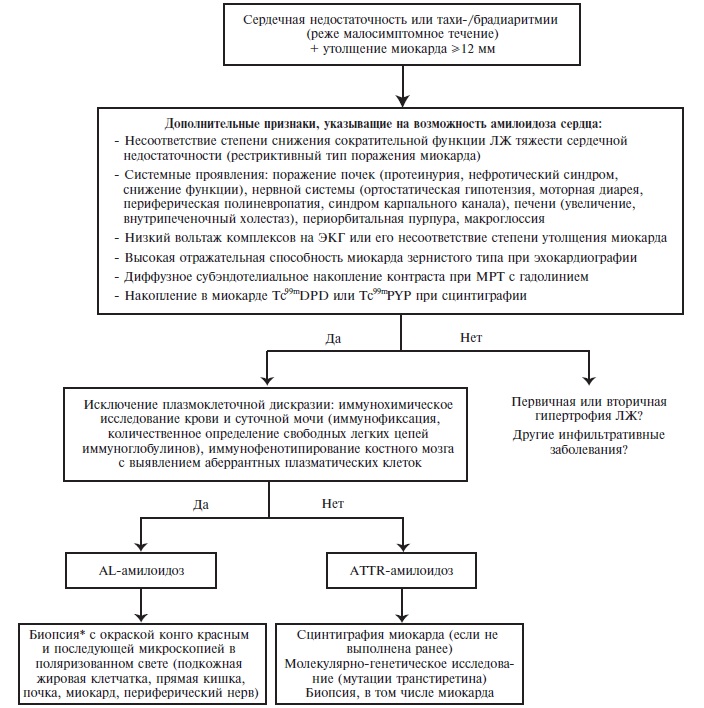 Рис. 1. Алгоритм дифференциальной диагностики амилоидоза сердца. *Если предполагается амилоидоз сердца, то скрининговую биопсию (подкожной жировой клетчатки, слизистой оболочки прямой кишки) целесообразно выполнить параллельно с другими исследованиями. При отрицательном результате показана биопсия пораженного органа, включая сердце.
Рис. 1. Алгоритм дифференциальной диагностики амилоидоза сердца. *Если предполагается амилоидоз сердца, то скрининговую биопсию (подкожной жировой клетчатки, слизистой оболочки прямой кишки) целесообразно выполнить параллельно с другими исследованиями. При отрицательном результате показана биопсия пораженного органа, включая сердце.
При AL-амилоидозе и особенно ATTR-амилоидозе часто встречается ортостатическая артериальная гипотензия – вариант сосудистой недостаточности, при которой сосуды теряют способность поддерживать нормальное артериальное давление в условиях ортостатических нагрузок. Она проявляется ощущением дурноты и потемнением в глазах в ортостазе в сочетании с резким снижением АД. Обычно этот симптом связан с дисфункцией автономной нервной системы (амилоидоз нервных сплетений сосудов). Тяжелая ортостатическая гипотензия сопровождается обмороками, а иногда приводит к развитию острого нарушения мозгового кровообращения.
Поражение желудочно-кишечного тракта может проявляться, особенно при AL-амилоидозе, тяжелой диареей или динамической непроходимостью, которые чаще связаны с нарушениями моторики кишечника вследствие дисфункции автономных нервных сплетений. Иногда выявляют изъязвления или перфорацию стенок с возможным кровотечением. При поражении пищевода возможна дисфагия.
Поражение печени при АА- и AL-типах амилоидоза наблюдают практически в 100% случаев. Функция печени чаще остается сохранной, редким признаком амилоидоза печени является внутрипеченочная портальная гипертензия. При некоторых вариантах семейного ALys-амилоидоза описаны тяжелые спонтанные внутрипеченочные кровотечения.
Увеличение селезенки, обусловленное амилоидным поражением, отмечается у большинства больных и обычно сопутствует увеличению печени.
Поражение нервной системы, представленное симптомами периферической соматической и автономной невропатии, отмечают у 17-35% больных AL-амилоидозом и практически у всех пациентов с наследственной амилоидной полиневропатией разных типов (ATTR, AApoA1 и др.). В большинстве случаев развивается дистальная симметричная полиневропатия с неуклонно прогрессирующим течением, различные дисфункции автономной нервной системы. Реже выявляют двусторонний синдром запястного канала, обусловленный сдавлением срединного нерва депозитами амилоида.
Поражение кожи наблюдают почти у 40% больных AL-амилоидозом. Помимо параорбитальных геморрагий описаны также папулы, бляшки, узелки, пузырьковые высыпания, склеродермоподобная индурация кожи.
Амилоидные отложения в мышцах чаще встречаются при AL-амилоидозе. Макроглоссия – патогномоничный симптом AL-амилоидоза, развивающийся примерно у 20% пациентов.
Редким проявлением амилоидоза, описанным при AL- и, в особенности, АTTR-типах, бывает поражение глаз (сухой кератоконъюнктивит, вторичная глаукома, помутнение стекловидного тела, дисфункции зрачка).
Клиническая картина других типов амилоидоза варьируется в зависимости от основной локализации и распространенности амилоидных депозитов, которые иногда могут быть значительными и напоминать проявления AL-амилоидоза.
Рекомендации:
Лечение системного амилоидоза
Целью терапии любого типа амилоидоза служит уменьшение количества (или, если возможно, удаление) белков-предшественников для того, чтобы замедлить или приостановить прогрессирование болезни. Неблаго приятный прогноз при естественном течении амилоидоза оправдывает применение агрессивных методов лечения. Клиническое улучшение, достигаемое с помощью лечения, включает стабилизацию или восстановление функции жизненно важных органов, а также предотвращение функциональных нарушений с увеличением продолжительности жизни больных. Лечение амилоидоза должно включать симптоматические методы, направленные на уменьшение выраженности сердечной недостаточности, аритмии, отечного синдрома, коррекцию артериальной гипотензии и др.
Лечение АА-амилоидоза
Цель терапии АА-амилоидоза – подавление продукции белка-предшественника SAA (вплоть до устойчивой нормализации), что достигается активным лечением хронического воспаления (в том числе субклинического). Это позволяет уменьшить клинические проявления и предотвратить прогрессирование амилоидной нефропатии и существенно улучшить прогноз.
Рекомендации:
Лечение АL-амилоидоза
При AL-амилоидозе, как и при множественной миеломе, целью лечения служит подавление пролиферации клона плазматических клеток для уменьшения продукции легких цепей иммуноглобулинов. В отличие от множественной миеломы, принципиальной задачей лечения AL-амилоидоза является по возможности полная элиминация патологического клона. В связи с быстрым прогрессированием заболевания важное значение имеет применение быстродействующих схем лечения на основе бортезомиба. По мере достижения ремиссии у некоторых больных применяют высокодозную химиотерапию с поддержкой аутологичными стволовыми клетками. При строгом подборе больных с исключением противопоказаний к этой терапии эффект достигают у 60% больных. У больных с клиническими симптомами амилоидоза сердца, ортостатической гипотензией, диареей, желудочно-кишечными кровотечениями в анамнезе, а также у лиц старше 70 лет с амилоидным поражением двух и более систем организма проведение высокодозной химиотерапии не рекомендуется. Тяжелый агранулоцитоз и другие осложнения существенно ограничивают ее применение. Проводят также лечение талидомидом или леналидомидом. Колхицин при AL-амилоидозе не эффективен.
Рекомендации:
Лечение ATTR-амилоидоза
До недавнего времени единственным методом лечения ATTR-амилоидоза была трансплантация печени, секретирующей нормальный транстиретин. Поскольку 98% всего сывороточного транстиретина синтезируется печенью, это позволяло прервать продукцию мутантного транстиретина. Трансплантация печени существенно замедляет прогрессирование ATTR-амилоидоза, а 20летняя выживаемость больных после трансплантации составляла 55,3% [36]. Однако уже имеющиеся массы амилоида способны выступать в роли ядра нуклеации для новых депозитов амилоида на основе нормального транстиретина (амилоидускоряющая субстанция). В настоящее время у больных с ранними стадиями ATTRамилоидной полиневропатии апробированы консервативные методы стабилизации тетрамерной структуры мутантного транстиретина и, следовательно, подавления его амилоидогенности. Один из таких препаратов – тафамидис замедлял на 52% (р=0,027) прогрессиро вание неврологических нарушений у больных ATTRамилоидозом, сохраняя функцию периферических соматических и автономных нервных волокон [12]. В клиническом исследовании III фазы лечение тафамидисом по сравнению с плацебо у больных с ATTR-амилоидозом сердца вызывало снижение общей смертности и частоты госпитализаций по сердечно-сосудистым причинам и задерживало ухудшение функциональной активности [81]. В Российской Федерации применение тафамидиса зарегистрировано только для лечения ATTR-амилоидоза с периферической полиневропатией.
В качестве стабилизатора транстиретина изучается также дифлюнизал из группы нестероидных противовоспалительных препаратов, однако его эффективность показана только в экспериментальных условиях.
СИСТЕМНЫЙ АМИЛОИДОЗ: диагноз, дифференциальный диагноз, лечение
«Амилоидоз» — термин, объединяющий группу заболеваний, которые отличаются большим разнообразием клинических проявлений и характеризуются внеклеточным отложением нерастворимых патологических фибриллярных белков в органах и тканях. Впервые эта патология бы
«Амилоидоз» — термин, объединяющий группу заболеваний, которые отличаются большим разнообразием клинических проявлений и характеризуются внеклеточным отложением нерастворимых патологических фибриллярных белков в органах и тканях. Впервые эта патология была описана в XVII в. Боне — саговая селезенка у больного с абсцессом печени. В середине XIX в. Вирхов применил ботанический термин «амилоид» (от греч. amylon — крахмал) для описания внеклеточного материала, обнаруженного в печени при аутопсии, так как полагал, что он близок по структуре к крахмалу. Впоследствии была установлена белковая природа отложений, однако термин «амилоид» сохранился до настоящего времени.
В 20-е гг. XX столетия Бенхольд предложил окрашивать амилоид конго-красным, затем был обнаружен эффект двойного лучепреломления в поляризованном свете — изменение кирпично-красной окраски на яблочно-зеленую. В 1959 г. Коген и Калкинс с помощью электронной микроскопии установили фибриллярную структуру амилоида.
Эволюцию претерпели и клинические представления об амилоидозе: Рокитанский в 1842 г. установил связь «сальной болезни» с туберкулезом, сифилисом, риккетсиозами; Уилкс в 1856 г. описал «жирные органы» у больного, не имевшего никаких сопутствующих заболеваний; Аткинсон в 1937 г. обнаружил амилоидоз у пациентов с миеломной болезнью. Выделены были старческие (Сойка, 1876) и наследственные (Андраде, 1952) формы заболевания, амилоидоз разделяли на генетический, первичный и вторичный, и, наконец, в 1993 г. была принята классификация ВОЗ, построенная на специфичности основного фибриллярного белка амилоида.
В нашей стране большой вклад в развитие представлений об амилоидозе внесли Е. М. Тареев, И. Е. Тареева, В. В. Серов. Огромная роль в изучении первичного и генетических вариантов амилоидоза и периодической болезни принадлежит О. М. Виноградовой, чьи монографии, изданные в 1973 и 1980 гг., не утратили своей актуальности и в наши дни.
В настоящее время амилоидоз принято клинически разделять на системные и локальные формы. Среди системных форм, в зависимости от состава фибриллярных отложений, выделяют четыре типа (табл. 1).
К локальным формам амилоидоза в настоящее время относят болезнь Альцгеймера (A-бета, фибриллы состоят из β-протеина, откладывающегося в головном мозге), амилоидоз островков поджелудочной железы, возможно, имеющий патогенетическую связь с диабетом 2 типа, амилоидоз, возникающий в эндокринных опухолях, амилоидные опухоли кожи, назофарингеальной области, мочевого пузыря и другие редкие виды.
AL-амилоидоз
Развитие AL-амилоидоза возможно при миеломной болезни, болезни Вальденстрема, В-клеточных лимфомах, и оно может быть идиопатическим при первичном амилоидозе. Все эти варианты объединены общим патогенезом, первичный амилоидоз представляет наибольшую трудность для распознавания в связи с отсутствием явных признаков гематологического заболевания, поэтому именно на данной форме стоит остановиться подробно.
При первичном амилоидозе, доброкачественной плазмоклеточной дискразии, родственной множественной миеломе, аномальные клоны плазматических клеток костного мозга продуцируют амилоидогенные иммуноглобулины. Некоторые аминокислоты в вариабельных участках легких цепей этих иммуноглобулинов занимают необычную позицию, что приводит к их нестабильности и обусловливает склонность к фибриллогенезу. У больных с первичным амилоидозом содержание плазматических клеток в костном мозге повышено до 5—10% (в норме их менее 4%, при миеломной болезни — более 12%), и они продуцируют определенный изотип легких цепей иммуноглобулинов, преобладающий при иммуногистохимическом окрашивании. Свободные моноклональные легкие цепи преобладающего лямбда- или (реже) каппа-изотипа определяются в крови и в моче, но содержание их ниже, чем при миеломной болезни.
Клиническая картина первичного амилоидоза многообразна и определяется преимущественным вовлечением в патологический процесс тех или иных органов — сердца, почек, нервной системы, желудочно-кишечного тракта, печени и др. Первыми симптомами являются слабость и потеря веса, но на этой стадии, до появления органных симптомов, диагноз устанавливается крайне редко.
Органами-мишенями при AL-амилоидозе чаще всего становятся почки и сердце. Поражение почек проявляется нефротическим синдромом, персистирующим и при наступлении ХПН, гематурия и артериальная гипертензия не характерны.
При отложении амилоида в миокарде развиваются разнообразные нарушения ритма, прогрессирующая сердечная недостаточность, чему могут предшествовать бессимптомные изменения на ЭКГ в виде снижения вольтажа зубцов. Эхокардиографическое исследование выявляет концентрическое утолщение стенок левого и правого желудочков, уменьшение объема полостей сердца, умеренное снижение фракции выброса, диастолическую дисфункцию миокарда левого желудочка.
Часто отмечаются симптомы вовлечения нервной системы — вегетативной, в виде ортостатической гипотензии, и периферической — в виде расстройств чувствительности. В последние годы стали описывать также поражения ЦНС, хотя ранее считалось, что они не характерны для первичного амилоидоза.
Диспептические явления (ощущение переполнения, запоры, поносы) и синдром нарушенного всасывания могут быть обусловлены как поражением вегетативной нервной системы, так и амилоидозом желудочно-кишечного тракта. Очень характерна гепатомегалия, природу которой следует дифференцировать между застойными явлениями вследствие сердечной недостаточности и амилоидным поражением печени. Последнее подтверждается повышением уровня щелочной фосфатазы сыворотки крови. Селезенка поражается часто, однако спленомегалия обнаруживается не всегда и большого клинического значения не имеет.
Макроглоссия, классический признак первичного амилоидоза, отмечается у 20% пациентов, инфильтрация мягких тканей может приводить к атрофии мышц, кожи, дистрофии ногтей, алопеции и появлению опухолевидных образований — амилоидом.
Реже встречается поражение сосудов, симптомами которого являются периорбитальная пурпура — «глаза енота» и экхимозы. Могут наблюдаться кровотечения, в том числе мочепузырные, обусловленные как изменением сосудистой стенки, так и нарушением свертывающей системы, в первую очередь дефицитом X-фактора, который связывается с амилоидом. Дефицитом факторов свертывания принято объяснять и характерный для амилоидоза тромбоцитоз.
Амилоидоз легких часто обнаруживается лишь при аутопсии. Однако в некоторых случаях одышка, кровохарканье и гидроторакс могут быть обусловлены не только застойной сердечной недостаточностью и нефротическим синдромом, но и амилоидным поражением легких. Возможны отложение амилоида в альвеолах и развитие легочных амилоидом. Рентгенологически могут выявляться сетчатые и нодулярные изменения в легочной ткани.
Поражение надпочечников может привести к надпочечниковой недостаточности, нередко остающейся нераспознанной, так как гипотензия и гипонатриемия рассматриваются как симптомы сердечной недостаточности и поражения вегетативной нервной системы. У 10—20% больных может иметь место гипотиреоз как проявление поражения щитовидной железы, нередко встречается увеличение подчелюстных слюнных желез.
Диагноз первичного амилоидоза помимо указанных клинических черт, которые могут быть сходными и при вторичном амилоидозе, базируется на ряде лабораторных данных. У 85% пациентов при иммуноэлектрофорезе белков сыворотки крови и мочи выявляются моноклональные иммуноглобулины. При рутинных исследованиях те же моноклональные иммуноглобулины обнаруживаются в моче в виде белка Бенс-Джонса. Биопсия костного мозга позволяет провести дифференциальный диагноз с множественной миеломой, а также выявить умеренное повышение количества плазматических клеток и их моноклональность при иммуногистохимическом окрашивании.
Однако даже сочетания характерной клинической картины и наличия моноклональных плазмоцитов и белков еще недостаточно для подтверждения диагноза первичного амилоидоза. Решающую роль здесь играют данные биопсии. Наименее инвазивной является аспирация подкожной жировой клетчатки передней брюшной стенки, дающая 80—90% положительных результатов при AL-амилоидозе (в нашей стране этот метод пока не нашел применения). Определенное диагностическое значение имеет биопсия десны и слизистой оболочки прямой кишки, но процент положительных результатов широко варьирует, в зависимости от стадии процесса, поэтому целесообразно выполнение биопсии одного из пораженных орга-нов — почки, печени, сердца, дающее почти 100% положительных результатов при амилоидозе AL-типа.
В первую очередь биопсийный материал окрашивается конго-красным. При обнаружении конгофилии исследуемого материала необходимо его исследование в поляризованном свете, эффект двойного лучепреломления характерен только для амилоида, другие конгофильные вещества яблочно-зеленой окраски не приобретают. После этого желательно типирование амилоида. Наиболее точным является иммуногистохимический метод с использованием моноклональных антител к белкам-предшественникам амилоида. Однако в настоящее время в нашей стране он практически недоступен. Поэтому для диагностики используется окраска с помощью растворов щелочного гуанидина или перманганата калия, что позволяет, хотя и косвенно, определить тип фибриллярных отложений.
Прогноз при первичном амилоидозе хуже, чем при других формах заболевания, средняя продолжительность жизни не превышает двух лет, при наличии поражения сердца или мультисистемного поражения без лечения больные погибают в течение нескольких месяцев. Наиболее частыми причинами смерти являются сердечная и почечная недостаточность, сепсис, сосудистые осложнения и кахексия. Патогенетическое сходство с миеломной болезнью позволяет рассчитывать на торможение прогрессирования заболевания при химиотерапии, проводимой с целью подавления моноклональных плазмоцитов. Существует несколько схем лечения (табл. 2).
Применение химиотерапии в случае успеха лечения позволяет увеличить продолжительность жизни больных на срок от 10 до 18 мес. Но эффективность терапии невысока, в частности, в связи с тем, что во многих случаях прогрессирование заболевания приводит к гибели больных до завершения курса лечения, а также из-за развития цитопении, инфекционных осложнений, фатальных нарушений ритма при лечении сверхвысокими дозами дексазона. Применение высоких доз мельфолана с трансплантацией аутологичных стволовых клеток позволяет достичь ремиссии более чем в 50% случаев, однако использование этого метода ограничено тяжестью состояния, возрастом больных, функциональными нарушениями со стороны сердца и почек. Во многих случаях возможна лишь симптоматическая поддерживающая терапия.
AA-амилоидоз
Развитие AA-амилоидоза происходит при хронических воспалительных процессах, предшественниками AA-амилоида являются сывороточные острофазовые белки, α-глобулины, продуцируемые клетками разных типов, в основном нейтрофилами и фибробластами. Вторичный амилоидоз развивается при ревматоидном артрите, болезни Бехтерева, псориатическом артрите, различных опухолях, лимфогранулематозе, неспецифическом язвенном колите и болезни Крона, при периодической болезни (семейной средиземноморской лихорадке), а также при туберкулезе, остеомиелите, бронхоэктатической болезни.
Характерными клиническими особенностями АА-амилоидоза является поражение почек у большинства пациентов, а также относительно редкое поражение печени и/или селезенки (около 10%) и сердца (выявляется лишь при эхокардиографии). Макроглоссия для вторичного амилоидоза не характерна. Диагноз основан на сочетании амилоидоза почек и хронического воспалительного заболевания, подтверждением служит иммуногистохимическое окрашивание биопсийного материала, в нашей стране используются уже упомянутые выше косвенные окрасочные методы.
Прогноз во многом зависит от природы основного заболевания, при естественном течении у трети больных через 5 лет от момента выявления протеинурии развивается почечная недостаточность. При периодической болезни пятилетняя выживаемость составляет 25%.
Лечение основано на подавлении очага — источника продукции сывороточных белков-предшественников. Удаление опухолей, секвестрэктомия, резекция кишки, лечение туберкулеза, уменьшение активности ревматоидного артрита (при использовании цитостатиков) приводят к прекращению прогрессирования амилоидоза, а иногда и к обратному развитию клинических проявлений, в частности нефротического синдрома.
Применение колхицина при периодической болезни является методом выбора, эффективность его доказана, лечение предотвращает развитие амилоидоза и тормозит его прогрессирование. При других формах вторичного амилоидоза эффективность колхицина не подтверждена.
Сенильные и наследственные формы системного амилоидоза, так же как и локальные формы, встречаются редко, диализный амилоидоз хорошо известен специалистам, в общей практике с ним сталкиваться практически не приходится.
Симптоматическая терапия зависит не от типа амилоидоза, а от пораженных органов-мишеней (табл. 3).
Амилоидоз, особенно первичный, считается нечастой патологией, однако в действительности он не столько редко встречается, сколько с трудом диагностируется. Адекватная диагностика требует не только знания клиники и патогенеза данного заболевания, но и наличия определенных диагностических возможностей. Чтобы проиллюстрировать это положение, приведем собственные данные (см. таблицу 4). В нефрологическом отделении МГКБ имени С. П. Боткина в 1993—2003 гг. наблюдалось 88 больных, которым был поставлен диагноз амилоидоза.
Диагноз был подтвержден морфологически у всех больных с AL-амилоидозом, старческим и неуточненным по типу амилоидозом, и у 30 пациентов со вторичным амилоидозом — всего в 53 случаях. У 12 больных выполнялась биопсия почки, а у двоих — биопсия печени, у восьми — биопсия кишки, в 12 случаях — десны, еще в 19 случаях диагноз был подтвержден при морфологическом исследовании секционного материала.
В большинстве случаев диагноз амилоидоза был установлен впервые в результате обследования в нефрологическом отделении. Нами было проведено сопоставление среди больных с AL-амилоидозом направительного и клинического диагнозов (табл. 5).
Лишь в двух случаях из 20 (10%) направительным диагнозом был «первичный амилоидоз», причем одному из этих больных он был поставлен в клинике терапии и профзаболеваний ММА, а другому — в зарубежной клинике.
Все больные, у которых диагностировалась миеломная болезнь с развитием AL-амилоидоза, были переведены в гематологические отделения. Из 11 больных с первичным амилоидозом семь пациентов получали химиотерапию комбинацией мельфолана с преднизолоном внутрь прерывистыми курсами, четверо из них — в сочетании с диализным лечением, и еще одна больная — только диализное и симптоматическое лечение. Из числа этих больных пять человек умерли в сроки от двух недель до двух лет от начала лечения (все с почечной недостаточностью и полиорганным поражением), один больной находится на диализе, один больной был направлен на трансплантацию аутологичных стволовых клеток, и одна больная получает лечение до настоящего времени. У одного пациента химиотерапия отложена в связи с наличием длительно не рубцующейся язвы желудка, и еще двое больных отказались от лечения.
Среди больных с вторичным амилоидозом в нашем исследовании преобладали пациенты с ревматоидным артритом, на втором месте среди причин — хронический остеомиелит и псориатический артрит, остальные заболевания встречались реже (табл. 6).
Лечение ревматоидного артрита и псориатического артрита проводилось с применением цитостатиков (метатрексата, азатиоприна), хотя во многих случаях возможности терапии были ограничены из-за наличия ХПН и сопутствующей патологии. Больные с хроническим остеомиелитом были направлены в отделения гнойной хирургии. Пациенты с болезнью Бехтерева и болезнью Крона получали специфическое лечение, больные с ХНЗЛ и туберкулезом также были направлены в профильные стационары. Одна из больных с опухолью желудка была успешно оперирована, и на протяжение четырех лет наблюдения нефротический синдром постепенно регрессировал, в остальных случаях опухолей распространенность процесса позволяла проводить только симптоматическую терапию, больной с лимфогранулематозом поступил в терминальном состоянии. Смертность среди пациентов со вторичным амилоидозом составила 38% (за счет больных с далеко зашедшим поражением на момент постановки диагноза). Все больные с периодической болезнью получали терапию колхицином.
Особенности диагностики и применения современных методов лечения первичного амилоидоза можно проиллюстрировать на следующем примере: больная К., 46 лет, впервые госпитализирована в конце октября 2002 г. с жалобами на отеки на ногах, сердцебиения, аменорею. В анамнезе — простудные заболевания, аппендэктомия, два нормальных срочных родоразрешения, указаний на заболевание почек, какие-либо хронические заболевания нет. В апреле 2002 г. перенесла острую пневмонию в верхней доле правого легкого, лечилась амбулаторно, получала инъекции абактала, линкомицина. В связи с локализацией пневмонии была обследована в туберкулезном диспансере, диагноз туберкулеза исключен. В начале июня впервые появились отеки на ногах, по поводу которых не обследовалась. Отеки через короткое время самостоятельно ликвидировались, затем возобновились. Больная была госпитализирована в терапевтический стационар, при обследовании выявлена протеинурия до 1,65%, гипопротеинемия (общий белок сыворотки крови 52 г/л), артериальное давление в норме (120/80 мм рт. ст.), мочевой осадок без изменений, креатинин плазмы также в пределах нормы. Установлен диагноз «острый гломерулонефрит», проведено лечение ампициллином, курантилом, гепарином, триампуром, выполнена тонзиллэктомия. Протеинурия сохранялась, отеки постепенно нарастали, в связи с чем для дальнейшего обследования и лечения больная с диагнозом «хронический гломерулонефрит» была направлена в больницу им. С. П. Боткина.
При осмотре — кожа чистая, обычной окраски, анасарка, отеки массивные, плотные, определяется асцит, периферические лимфатические узлы не увеличены. АД 110/70 мм рт. ст., тоны сердца звучные, ясные, ритмичные, ЧСС 90 уд/мин, печень и селезенка не увеличены, диурез до 1000 мл/сут, стул регулярный, без патологических примесей. При обследовании выявлен нефротический синдром — протеинурия 3 г/л, мочевой осадок скудный, гиподиспротеинемия, гиперлипидемия (общий белок сыворотки крови 39 г/л, альбумины 12 г/л, глобулины 7-30-15-19% соответственно α1-α2-β-γ холестерин 17,8 ммоль/л, β-липопротеиды 250 ЕД), при анализе мочи на белок Бенс-Джонса — реакция отрицательная, суточная экскреция 17-КС не снижена. Клинический анализ крови и другие биохимические показатели в пределах нормы, коагулограмма — выраженная гиперфибриногенемия, повышение уровня РКФМ. Исследование иммуноглобулинов крови: Ig-A — 0,35, Ig-M — 35,7 (две нормы), Ig-G — 1,96 г/л. Рентгенография органов грудной клетки, костей черепа и таза, УЗИ брюшной полости, почек, щитовидной железы, ЭХО-КГ без патологии, УЗИ малого таза — признаки аденомиоза тела матки, ЭГДС — рефлюкс-эзофагит, хронический гастрит. При осмотре невропатологом патологии не найдено, онкологом установлена фиброзно-кистозная мастопатия.
С целью уточнения генеза нефротического синдрома под местной анестезией под УЗ-наведением выполнена тонкоигольная пункционная биопсия правой почки, осложнений не было. При исследовании биоптата в мезангии клубочков и во внегломерулярных сосудах отмечается отложение амилоида. Амилоид загружает до 25% сосудистых петель клубочков. При иммуногистохимическом исследовании специфической люминисценции не найдено. При обработке препаратов раствором щелочного гуанидина в течение 2 ч конгофилия и их свойства в поляризованном свете сохраняются, что характерно для AL-амилоидоза.
Для выяснения природы AL-амилоидоза выполнено иммунохимическое исследование крови и мочи в лаборатории «Иммунотест». Выявлена М-лямбда парапротеинемия со снижением уровня поликлональных иммуноглобулинов и парапротеинурия Бенс-Джонса лямбда-типа на фоне массивной неселективной протеинурии. Больная была консультирована гематологом, высказано предположение о наличии болезни Вальденстрема, произведена трепанобиопсия костного мозга. Заключение: в имеющихся костно-мозговых полостях видны клетки всех трех ростков нормального гемопоэза, а также лимфоидные клетки, не образующие скоплений. Диагноз болезни Вальденстрема отвергнут в связи с отсутствием лимфоидной инфильтрации костного мозга, увеличения лимфоузлов и селезенки и отсутствием субстрата опухоли.
Установлен диагноз первичного амилоидоза с поражением почек, нефротическим синдромом, сохранной почечной функцией, признаков иных органных поражений не выявлено. С января 2003 г. начата химиотерапия мельфоланом 16 мг/сут и преднизолоном 100 мг/сут, курсами по четыре дня каждые шесть недель. Проводится также симптоматическое лечение: фуросемид, верошпирон, препараты калия, фамотидин, переливания альбумина. К настоящему времени проведено пять курсов химиотерапии с хорошей переносимостью, отеки уменьшились, протеинурия снизилась до 1,8 г/л, несколько уменьшилась выраженность гиподиспротеинемии (общий белок 46 г/л, альбумины 18 г/л, α2-глобулины 20%). Функция почек остается сохранной, креатинин плазмы 1,3 мг/Дл, признаков поражения других органов и систем при контрольных динамических обследованиях не выявлено.
Данный случай наглядно иллюстрирует тот факт, что для диагностики амилоидоза необходимо морфологическое, иммунологическое и иммунохимическое обследование. Так, у нашей пациентки наиболее очевидным клиническим диагнозом был «хронический гломерулонефрит», и в отсутствии возможности выполнения биопсии почки именно этот диагноз, скорее всего, и был бы поставлен. Никаких клинических указаний на системный характер заболевания, хронический воспалительный процесс, заболевание системы крови, за исключением повышения уровня Ig-M, у больной не было. И лишь полученные при исследовании почечного биоптата данные повлекли за собой трепанобиопсию костного мозга и иммунохимическое исследование, что в совокупности позволило поставить диагноз первичного амилоидоза до появления системных повреждений. Патогенетическая терапия была начата хотя и на фоне уже развившегося нефротического синдрома, но до наступления почечной недостаточности и при загрузке лишь 25% клубочков амилоидом, что прогностически относительно благоприятно.
В заключение отметим, что амилоидоз представляет собой тяжелое заболевание с высоким уровнем летальности, которое чрезвычайно трудно диагностировать, однако своевременное и качественное обследование больных позволяет поставить диагноз в более ранние сроки, а своевременное назначение адекватной терапии, в свою очередь, дает возможность улучшить прогноз в этой группе больных.
Литература
Е. В. Захарова
Московская городская клиническая больница им. С. П. Боткина