Синдром Ретта у детей: общие сведения и симптомы
 Синдром Ретта – это генетическое заболевание, сопровождающееся тяжелыми психоневрологическими симптомами. Диагностика его затруднительна: оно практически никогда не обнаруживается внутриутробно, а после рождения проявляется не ранее, чем через 6 месяцев. Своему носителю оно грозит глубоким слабоумием, двигательными ограничениями, дезадаптацией в социуме.
Синдром Ретта – это генетическое заболевание, сопровождающееся тяжелыми психоневрологическими симптомами. Диагностика его затруднительна: оно практически никогда не обнаруживается внутриутробно, а после рождения проявляется не ранее, чем через 6 месяцев. Своему носителю оно грозит глубоким слабоумием, двигательными ограничениями, дезадаптацией в социуме.
Истоки заболевания
В масштабном формате о расстройстве заговорили в 1983 году благодаря шведскому ученому Бенгту Хагбергу. В это время он со своей группой изучал 35 подобных между собой случаев в 3 разных странах: в Португалии, Франции и Швеции.
Однако Хагберт не является первооткрывателем синдрома. Впервые его обнаружил педиатр Андреас Ретт, имя которого носит заболевание. Он наблюдал за двумя девочками, имеющими одинаковые симптомы. Их он заметил в очереди на прием. Они сидели на коленях у матерей, а те держали их за руки. Девочки раскачивались как маятники, а затем внезапно обе начали совершать стереотипные движения руками. Дети застыли в одном положении, отстраненные от окружающего мира. Взгляд был направлен в одну точку. Поражала их синхронность в движениях и поведении.
В своих письменных архивах врач отыскал подобные истории болезни, а затем отправился в Европу, чтобы разыскать и там таких же пациентов. В 1966 он сделал первые публикации своих исследований, которые, однако, не вызвали особого интереса.
Зафиксированную им болезнь Ретт назвал синдромом атрофии мозга. Сначала ее считали проявлением аутизма или шизофрении, и только лишь в 1983 году вывели в отдельную нозологическую единицу.
В настоящее время синдром относят к категории довольно редких генетических заболеваний. Он встречается с частотой случаев 1 на 15000. Причиной его называют мутацию гена МЕСР2. Этот ген отвечает за синтез определенного белка, влияющего на развитие мозга. В норме этот белок, сп устя некоторое время после рождения, должен подавляться другими генами, чтобы обеспечить нормальное развитие мозга.
устя некоторое время после рождения, должен подавляться другими генами, чтобы обеспечить нормальное развитие мозга.
Если же ген МЕСР2 мутирован, то белок инактивируется не полностью, что вызывает аномальное мозговое созревание, и провоцирует развитие синдрома Ретта.
Обычно мутирующий ген располагается в Х хромосоме, потому заболеванием страдают преимущественно девочки.
Почему мальчики не болеют
Учитывая, что мутирующий ген несет в себе Х-хромосома, то девочки в плане заболевания находятся в более «выигрышной» позиции. У них присутствует две Х-хромосомы. Поэтому если одна из них «бракованная», то вторая функционирует нормально. Это дает девочке хоть малый шанс на нормальное существование.
У мальчика Х-хромосома одна. Если она имеет мутационный ген, значит, выпадает из работы полностью, и ее нечем заменить. Такие малыши мужского пола, как правило, погибают еще внутриутробно, так и не родившись. Поэтому синдром Ретта у мальчиков встречается крайне редко.
 Но, несмотря на такую особенность заболевания, очень редко, но все-таки мальчики с подобным синдромом выживают. Это может быть связано с тем, что не все гены в Х-хромосоме подвергаются мутации. Из-за этого заболевание развивается не столь остро.
Но, несмотря на такую особенность заболевания, очень редко, но все-таки мальчики с подобным синдромом выживают. Это может быть связано с тем, что не все гены в Х-хромосоме подвергаются мутации. Из-за этого заболевание развивается не столь остро.
Другая причина – наличие у мальчика синдрома Клайнфельтера. При этом наблюдается полисомия половых хромосом, то есть их набор составляет ХХУ. И, если одна Х-хромосома имеет патологический ген, то вторая может регулировать синтез белка и дарить мальчику возможность жизни. Получается такая же картина, как и у девочки.
Как развивается заболевание
Синдром Ретта у детей – довольно коварное заболевание. При рождении оно практически не проявляет себя. Первые его симптомы появляются в период от 6 мес. до полутора лет. Однако некоторые, еле заметные признаки, в первом полугодии все-таки имеются. Но они настолько ничтожны, что не привлекают внимания.
Вот что говорит мама одной из девочек с синдромом по поводу первого полугодия ее жизни. Она придала значение этим мелочам только по прошествии 1 года и 7 месяцев с рождения ее дочери, когда проявления стали уже явными. Из предвестников болезни она отметила, что ее малышка начала держать голову в 3 месяца, а не в 2, как это положено. В 6 месяцев она еще не могла сидеть, а ходить начала только в 1 год и 4 месяца. Психологически развивалась нормально, и говорить начала рано, но это были не стандартные слова «мама», «папа», а «зайчик», «мишка» и др.
В 1 год и 7 мес. она перестала узнавать родителей и, казалось, не нуждалась в них. Весь день проводила за одним однообразным занятием: кидала мяч или катала коляску. Часами ходила по кругу, пока ее не останавливали или она запиналась. Такое стереотипное поведение носит название полевого, когда действие затягивает больного, и он не может ничего сделать.
В четыре года к симптомам присоединились эпилептоидные припадки. Однако по достижении школьного возраста девочка находилась на домашнем обучении, и делала некоторые успехи. 
12–6 лет – это был период ремиссии, когда болезнь практически не беспокоила. Но с 16 лет появились новые, более глубокие проблемы, связанные с костными деформациями и болезнями внутренних органов. Одна нога девочки была короче другой почти на 10 см, что не могло не препятствовать ходьбе. В 20 лет она весила всего 24 кг с ростом 158 см.
Обычно СР протекает в 4 стадии.
Первая стадия, которая, как правило, стартует с 6 месяцев до полутора лет, проявляется нарастанием раздражительности и лабильностью настроения у ребенка. Эпизоды плача и психомоторного возбуждения сменяются все большей пассивностью. Малыш бесцельно передвигается по комнате, пропадает интерес к игрушкам. Но контакт с матерью сохраняется.
Вот как описывает женщина поведение своей дочери на заре заболевания: она кричала целый день без остановки, билась головой о стены, не могла уснуть. Что бы мы ни делали, она не успокаивалась. Это был настоящий ад. Но больше угнетало то, что ни один врач не мог поставить вразумительный диагноз.
Развивается диспропорция головы и конечностей по отношению к телу. Они становятся несоизмеримо маленькими. Замедляется рост, и снижается тонус мышц.
Вторая стадия, длящаяся несколько лет, отличается пестротой симптомов. Сразу обращает на себя внимание снижение интеллектуальных способностей, развивается умственное слабоумие. Происходит регресс практически всех полученных навыков. Речь полностью исчезает или переходит в степень эхолалии – механического повторения услышанного.
Приобретенные двигательные навыки, предметно-ролевое поведение теряются и замещаются двигательными стереотипами. Характерный симптом: многочисленно повторяющиеся движения, напоминающие мытье рук. Кроме этого, ребенок постоянно заламывает или потирает их, размахивает ими, хлопает в ладоши. Сжатие пальцев рук вполне нормально в 4 месяца, но в более позднем возрасте говорит об остановке развития. Малыш утрачивает хватательный рефлекс, не способен производить вращательные движения руками.
Постепенно двигательная активность сходит на нет. Нарушается походка, ребенок ходит, не сгибая коленей.
 Третья стадия длится 10 лет и более, характеризуется она развитием стойкого, глубокого слабоумия, вплоть до идиотии. Наблюдается полная потеря способности говорить и понимать обращенную к ребенку речь. Появляется тремор всего тела, отягчающий движения. Усиливаются судорожные припадки.
Третья стадия длится 10 лет и более, характеризуется она развитием стойкого, глубокого слабоумия, вплоть до идиотии. Наблюдается полная потеря способности говорить и понимать обращенную к ребенку речь. Появляется тремор всего тела, отягчающий движения. Усиливаются судорожные припадки.
Четвертая, конечная стадия – это период усугубления ранее проявляемых симптомов. Стойкая утрата умственных способностей, двигательных навыков, развитие мышечных дистрофий, приводящих к полному обездвиживанию.
Продолжительность жизни таких больных в среднем колеблется до 30 лет, хотя известны случаи, когда они доживали и до 50-летнего возраста.
Самые частые симптомы расстройства
Типичные симптомы для синдрома Ретта – мышечные и двигательные нарушения. Мышцы находятся в гипертонусе или же, наоборот, теряют его. В этом случае у ребенка развивается неправильное положение тела, прогрессирует частичные параличи и нарушение координации. Например, девочки скрещивают ноги во время ходьбы.
Синкинезии – патологические сокращения мышц, возникают вслед за произвольным движением: простая улыбка способна вызвать резкий взмах ногой. Такое явление постепенно приводит к повреждению суставов, сухожилий и связок, провоцирует ортопедические нарушения. Последние проявляются во всевозможных деформациях и также очень часто сопровождают таких детей. Среди них выделяют вывих тазобедренного сустава, провоцируемый малой подвижностью.
Статическая деформация стопы чаще развивается из-за нарушенного мышечного тонуса. Распространенной считается патология под названием «конская стопа», связанная со снижением подвижности голеностопного сустава. Ее можно узнать по пятке, которая не достигает земли, стопа при этом смещается кнаружи или вовнутрь. Причина патологии – гипертонус икроножной мышцы.
Сколиоз – боковое искривление позвоночника, который провоцирует массу проблем у таких пациентов: деформации суставов и костей, боли во время ходьбы, в стоячем или сидячем положении, утрата способности передвигаться. Сколиоз грудного отдела вызывает легочную недостаточность. Появляются также проблемы с пищеварением.
У детей с синдромом Ретта наблюдается повышенное слюнотечение. Но это происходит не из-за избытка количества слюны, а потери способности сглатывать ее.
Нарушение питания может развиваться из-за частых приступов тошноты. Она появляется на любые аспекты питания: на определенный продукт, его температуру, на способ приготовления. Так, ребенок способен отрицательно реагировать на пищу, поданную кусочками, или на комочки в блюде.
Постоянная тошнота провоцирует отказ от питания, а значит, потерю в весе. 
Плохое сглатывание слюны, которая регулирует кислотность в желудке, и повышенное внутрибрюшное давление вызывают желудочно-пищеводный рефлюкс, то есть забрасывание содержимого желудка в пищевод. Это чревато такими последствиями, как воспаление стенки пищевода, респираторные инфекции.
Малоподвижный образ жизни, неврологические расстройства, неправильное питание провоцируют возникновение запоров у детей с синдромом Ретта. Они носят тяжелый характер, поскольку способны вызывать закупорку кишечника и сильные боли.
Повышенное слюнотечение, тошнота, рефлюкс снижают потребление ребенком пищи и даже развивают на нее негативную реакцию. В результате этого ребенок теряет в весе. Этот процесс стоит строго контролировать, поскольку он чреват истощением.
Другое тяжелое расстройство связано с работой дыхательной системы, развивающееся вплоть до приступов апноэ. Это явление настолько часто среди детей с синдромом, что нередко стает причиной их гибели.
Важными патогномоничными признаками синдрома считаются проявления аутизма. Именно из-за них заболевание изначально считали одной из форм этого расстройства, а в настоящее время относят к болезням аутистического спектра.
Аутистические признаки проявляются в отстранении от окружающего мира, в том числе и от родственников. Ребенок замыкается в себе, может не откликаться, когда его зовут. Предпочитает одиночество. Дети боятся чужих людей и непривычных ситуаций.
Лицо такого ребенка становится похожим на каменное. Взгляд блуждающий или устремлен в одну точку. Поведение часто непредсказуемо: случаются приступы неутомимого смеха или плача. Склонны к самоповреждениям: царапают кожу, кусают пальцы, вырывают волосы.
Нетипичная картина
Наряду с типичной формой заболевания, описанной выше, встречаются и атипичные формы. Они имеют свои особенности, от которых зависит тяжесть заболевания.
 Синдром Ретта – сложное генетическое заболевание. Прежде всего, его сопровождает полная умственная деградация и психоневрологические нарушения, влекущие за собой многочисленные патологии других систем организма.
Синдром Ретта – сложное генетическое заболевание. Прежде всего, его сопровождает полная умственная деградация и психоневрологические нарушения, влекущие за собой многочисленные патологии других систем организма.
К сожалению, в мире еще не существует способа кардинального искоренения болезни, хотя ученые ведут постоянные разработки в этом направлении.
Лечение синдрома сводится к трем основным направлениям. Медикаментозная терапия назначается для купирования судорожных припадков и стимуляции работы головного мозга.
Диетотерапия включает в себя контроль массы тела, употребление в пищу высококалорийных, витаминизированных продуктов.
Однако наибольшее внимание уделяется реабилитационным мероприятиям, направленным на укрепление опорно-двигательного аппарата и поддержание умственного, психомоторного развития.
Важно сохранить комплексный, всесторонний подход к проблеме. Такие дети нуждаются в постоянной поддержке со стороны взрослых и веры в них. Сотрудничество с ними, как с полноценной ячейкой общества, способствует их лучшей адаптации в социуме и более благоприятному развитию.
Синдром Ретта
Синдром Ретта – генетическое заболевание, характеризующееся нарушением развития нервной системы по причине отсутствия ингибирования определенных генов. Проявлениями этого состояния являются прогрессирующая умственная отсталость у девочек (при крайне редких атипичных формах – и у мальчиков), мышечная гипотония, атаксия, искривления позвоночника. Диагностика синдрома Ретта основывается на данных общего и неврологического осмотра, магнитно-резонансной томографии, электроэнцефалографии и молекулярно-генетических анализов. Специфического лечения не существует (имеются лишь определенные наработки с обнадеживающими результатами при опытах на животных), применяют симптоматическую терапию для облегчения состояния больного.
Общие сведения
Синдром Ретта – генетическое заболевание психоневрологического характера, практически всегда развивающееся у девочек и проявляющееся тяжелой степенью умственной отсталости. Эта патология была впервые выявлена еще в 1954 году австрийским неврологом А. Реттом, однако в качестве отдельной нозологической единицы он выделил данное заболевание лишь в 1966 году. Широкую известность в научном мире синдром Ретта получил в 1983 году после исследований Б. Хагберга. Это состояние является довольно распространенным – его встречаемость составляет примерно 1:10-15 тысяч новорожденных девочек, всего на сегодняшний день описано несколько десятков тысяч случаев патологии. Механизм наследования синдрома Ретта – доминантный, сцепленный с Х-хромосомой, именно поэтому он встречается практически всегда у девочек. У мальчиков из-за отсутствия парной Х-хромосомы генетические повреждения, приводящие к такому заболеванию, почти всегда являются летальными. Однако существует несколько атипичных форм синдрома Ретта, характеризующихся более сглаженной клинической картиной и поэтому поражающих лиц мужского пола. Кроме того, у мальчиков такая патология может развиться при наличии дополнительной Х-хромосомы – синдроме Клайнфельтера.
Причины синдрома Ретта
Этиология и патогенез синдрома Ретта достаточно сложны и обусловлены взаимодействием различных генов и их влиянием на развитие головного мозга человека. Первопричиной заболевания является нонсенс-мутация (по некоторым данным, к аналогичным нарушениям приводят и миссенс-мутации) гена MECP2, локализованного на Х-хромосоме, в результате чего его экспрессия полностью прекращается. Он кодирует специфический протеин под названием метил-CpG-связывающий белок 2, участвующий в регуляции транскрипции определенных участков ДНК. Данный белок содержит два домена, один из которых способствует его присоединению к метилированным участкам хромосом (которые расположены вблизи генов, регулирующих развитие головного мозга), а второй выступает как репрессор транскрипции. Причина синдрома Ретта как раз и заключается в отсутствии ингибирования некоторых генов, что приводит к нарушению формирования нервной ткани.
При этом синдром Ретта нельзя рассматривать как нейродегенеративное заболевание, так как при нем не наблюдается разрушения нейронов или нейроглии. Гистологические исследования тканей головного мозга больных выявляют нарушение ультраструктуры нервных клеток – уменьшение размеров, изменение количества дендритов, затрудненное образование нервных тканей. Объем нейроглии при синдроме Ретта снижен, в результате этого на макроскопическом уровне размер головного мозга тоже уменьшается на 20-30% по сравнению с возрастной нормой. Одной из причин вышеперечисленных процессов является отсутствие торможения выделения фермента GAD67 (ингибирование гена этого энзима осуществляется метил-CpG-связывающим белком 2), что, в свою очередь, приводит к увеличению концентрации тормозных трансмиттеров из группы ГАМК. В результате этого у больных синдромом Ретта наблюдается значительное превалирование процессов торможения в головном мозге, что отражается не только на физиологии центральной нервной системы, но и на ее морфологическом строении.
Врачами-генетиками было установлено, что полное отсутствие в геноме нормального гена MECP2 в подавляющем большинстве случаев является летальным состоянием и нередко приводит к внутриутробной смерти плода. Такое состояние имеет место у мальчиков (по причине наличия только одной Х-хромосомы) или у девочек-гомозигот, что встречается крайне редко. Из-за этого в половом распределении синдрома Ретта наблюдается абсолютное превалирование больных женского пола. Мутации гена MECP2 в большинстве случаев являются спонтанными или герминативными – предположительно, 70% случаев этого заболевания обусловлено генетическим дефектом Х-хромосомы в половых клетках отца. Дефекты этого гена приводят и другим патологиям центральной нервной системы – варианту Запела, синдрому Луба (Х-сцепленная умственная отсталость у мальчиков), врожденной энцефалопатии. Некоторые исследователи относят эти состояния к атипичным формам синдрома Ретта.
Симптомы синдрома Ретта
У новорожденных девочек синдром Ретта поначалу никак не проявляется, первые 6-12 месяцев развитие ребенка происходит обычными темпами без каких-либо отклонений. В дальнейшем прогрессирование заболевания характеризуется определенной стадийностью. Первая стадия синдрома Ретта, чаще всего возникающая в возрасте от 6-ти месяцев до 2,5 лет, характеризуется появлением у ребенка мышечной гипотонии, замедления психомоторного развития с последующим отставанием от сверстников, потерей интереса к играм и окружающим людям. Врачи-педиатры отмечают более медленный, нежели в норме, рост стоп и кистей в длину и замедление роста окружности головы. Иногда помимо неврологических проявлений может отмечаться нарушение работы печени, сердца, желудочно-кишечного тракта.
Вторая стадия синдрома Ретта характеризуется более выраженными клиническими проявлениями. Она развивается на протяжении 1-2 лет после появления первых симптомов заболевания, при этом у ребенка сначала наблюдается беспокойство, нарушения сна. Затем довольно быстро, всего за несколько недель, больные синдромом Ретта теряют практически все приобретенные до этого времени навыки – утрачивается речь, исчезает способность к ходьбе. Также для этой стадии развития патологии характерны расстройства дыхания – периоды апноэ по 1-2 минуты могут перемежаться с приступами учащенных и глубоких дыхательных движений (гипервентиляция). Дыхательные нарушения при синдроме Ретта отличаются наличием только при бодрствовании больного и отсутствием во время сна. Часто возникают многочисленные неврологические нарушения: атаксия, эпилептические припадки, часто повторяющиеся стереотипные движения.
Третья стадия синдрома Ретта называется псевдостационарной, так как при ней мало заметны признаки прогрессирования заболевания. Обычно она длится от 4 до 15 лет, состояние больных стабильно, однако наблюдаются судорожные приступы, глубокая умственная отсталость, гиперкинезы. В большинстве случаев синдрома Ретта этот этап оканчивается в пубертатном периоде. Четвертая стадия синдрома Ретта характеризуется уменьшением частоты эпилептических припадков вплоть до их исчезновения при прогрессировании двигательных расстройств. Большинство больных полностью теряют подвижность, возникает атрофия мышц, сосудистые нарушения в нижних конечностях, что может привести к развитию трофических язв. Из-за слабости мышечного корсета спины при синдроме Ретта возникает сколиоз или другие формы искривления позвоночника.
Диагностика
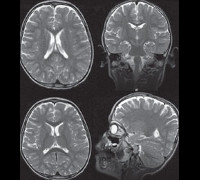
Диагностика синдрома Ретта производится на основании изучения анамнеза больного, его настоящего статуса, магнитно-резонансной томографии и энцефалографии, молекулярно-генетических анализов. Изучение наследственного анамнеза, как правило, не имеет особого смысла по причине спорадического характера мутаций гена MECP2. Характерными для синдрома Ретта являются нормальное развитие ребенка до 6-12 месяцев, возникновение мышечной гипотонии и беспокойства в раннем детстве, появление в дальнейшем атаксии и частых эпилептических припадков, стремительное утрачивание приобретенных навыков. В дальнейшем у больных регистрируется тяжелая умственная отсталость, мышечная слабость (вплоть до атрофии), искривление позвоночника, судорожные припадки.
При осмотре больных синдромом Ретта выявляется отставание в росте и его остановка, резкое уменьшение окружности головы, отсутствие речи (на начальных этапах патологии характерна эхолалия). На магнитно-резонансной томографии головного мозга обнаруживается уменьшение размера органа, нечеткая дифференциация серого и белого вещества, базальных ганглиев, снижение складчатости коры больших полушарий. Электроэнцефалограмма подтверждает снижение фоновой активности головного мозга и резко ослабленную реакцию на внешние раздражители.
Наиболее точную диагностическую информацию дают методы современной генетики – поиск делеций в локусе гена MECP2 или прямое секвенирование его последовательности для определения мутаций. Такое подтверждение синдрома Ретта возможно и в рамках пренатальной диагностики генетических заболеваний. Вспомогательную роль в установлении этого состояния может играть обследование внутренних органов (например, методами УЗИ) – у 20-30% больных выявляется недоразвитие печени или селезенки.
Лечение синдрома Ретта
Специфического лечения синдрома Ретта на сегодняшний день не существует. Имеются обнадеживающие данные некоторых исследовательских лабораторий, сотрудникам которых удалось «включить» ген MECP2 у мышей и тем самым добиться исчезновения симптомов заболевания. В сфере практической медицины пока доступна только симптоматическая терапия, однако и она сопряжена с рядом трудностей – в частности, эпилептические припадки при этом заболевании плохо поддаются устранению противосудорожными средствами. Также для лечения синдрома Ретта применяют ноотропные препараты, нарушения сна корректируют снотворными препаратами из группы барбитуратов или мелатонином.
Прогноз и профилактика
«Я просто хочу что-то сделать для таких детей, как моя дочь»
Как живут семьи, воспитывающие детей с редкой генетической мутацией
Каждый год в мире увеличивается количество детей и взрослых с редкими заболеваниями. Продолжительность их жизни зависит от своевременной диагностики и качества ухода и реабилитации. Режиссер Мария Иванова-Сурае, глава Ассоциации семей, воспитывающих детей с мутацией в гене CDKL5, рассказала спецкорреспонденту “Ъ” Ольге Алленовой, какая государственная помощь необходима детям с таким заболеванием, зачем нужны гранты для российских генетиков и почему, по ее мнению, органы социальной защиты не выполняют свою основную задачу.

Мария долгое время не хотела принять диагноз дочери и пыталась ее вылечить. Только когда Таисии исполнилось пять лет, ее мать приняла болезнь и стала улучшать качество жизни ребенка
Фото: Архив Марии Ивановой-Сурае
Мария долгое время не хотела принять диагноз дочери и пыталась ее вылечить. Только когда Таисии исполнилось пять лет, ее мать приняла болезнь и стала улучшать качество жизни ребенка
Фото: Архив Марии Ивановой-Сурае
Долгое время детям с мутацией в гене CDKL5 диагностировали синдром Ретта, и только в 2012 году было выявлено отдельное заболевание. Мутация CDKL5 приводит к ранней детской эпилепсии, ДЦП, расстройствам аутистического спектра, умственной отсталости. В мире сегодня с этим диагнозом живет более 1 млн детей. С мутацией такого гена рождается один ребенок на 40 тысяч. В России заболевание официально диагностировано у 65 детей. На самом деле их больше, но в регионах диагностировать болезнь пока не научились.
В 2019 году в России была создана Ассоциация CDKL5 Russia, возглавила ее продюсер и режиссер Мария Иванова-Сурае, воспитывающая ребенка с таким заболеванием. Ассоциация входит в состав международной организации CDKL5 Alliance.
«Такие дети живут в своем мире, они себя прячут очень глубоко»
— Как быстро поставили диагноз вашей дочери?
— Я с 2006 года снимаю документальное кино, а в 2008-м сняла фильм про детей с аутизмом. Вскоре после этого у меня родилась Таисия. До ее двух месяцев я ничего особенного не замечала, она первый ребенок, опыта у меня нет. Но потом я поняла, что она не следит за предметами, не концентрирует взгляд, не узнает ни меня, ни отца, не обращает внимание на внешние раздражители. И дочка была какая-то вялая. Когда ей было два месяца, у нее случился эпилептический приступ, мы вызвали скорую, нас доставили в Морозовскую больницу. Там начали применять противосудорожную терапию, но ей это не помогало — приступы случались почти каждый день. Мы перепробовали самые разные варианты лечения, но состояние Таисии не улучшалось.
Мы не знали, что делать. Моя подруга рассказала, что в Германии живет доктор Ханс Хольтхаузен, он уже тогда был в преклонном возрасте, а сейчас ему 90 лет. И Хольтхаузен, увидев фотографию моей дочери, видеозапись ее приступа и прочитав описание ее состояния, предложил нам сделать анализ на мутацию гена CDKL5. В России тогда такой анализ сделать было нельзя. Мы поехали в Мюнхен, диагноз подтвердился. Мы с отцом Таисии тоже сдавали анализы, но оказалось, что у нас обоих нет мутации этого гена — врачи в Мюнхене объяснили нам, что мутация спонтанная. Она непредсказуемая. Сейчас в мире один из 40 тысяч детей рождается с такой мутацией.
Врачи тогда сказали нам, что это очень тяжелые дети, они редко живут более семи лет, и они не будут говорить, ходить, стоять. Я это не приняла. С отцом Таисии мы расстались.
В России есть знаменитый врач — профессор Константин Мухин,— который занимается эпилепсией. Мы к нему пришли на прием, когда Таисии было полтора года. Он, когда ее увидел, сразу сказал, что она очень тяжелый ребенок. Но я все равно отказывалась в это верить. И продолжала бороться.
Я сутками сидела на форумах, читала про каких-то целителей. Помню, вычитала про деревенскую бабку, которая чудеса творит. Мы с Таисией сели в машину, ехали сутки, приезжаем, а она посмотрела на нас и говорит: «Ой, а я лечу только лишай». Мы в каком-то городишке переночевали и поехали обратно.

Как пациенты с редкими болезнями добиваются помощи, гарантированной им государством
Дочь наблюдается в НПЦ в Солнцево (Научно-практический центр специализированной медицинской помощи детям имени В. Ф. Войно-Ясенецкого.— “Ъ” ), ее лечащий врач, заведующая отделением неврологии и эпилептологии, предложила нам курс гормонотерапии, я согласилась,— это лечение дало Таисии ремиссию на полгода, она стала развиваться, даже пробовала сидеть. О том, чтобы ходить, не было и речи, но по крайне мере я заметила подвижки, и это дало мне силы. Как-то я узнала, что на Украине есть доктор, который ставит детей с ДЦП на ноги, таких случаев у него было много, я видела эти видео, общалась с родителями, которые возили к нему детей. У него своя методика, он мягко работает руками, как остеопат, но он ею ни с кем не делится. Противопоказание для лечения — эпилепсия, и я долго думала, стоит ли рисковать, но в итоге решилась. Курс стоил дорого, я обратилась в фонд Гоши Куценко «Жизнь с ДЦП», и они помогли нам оплатить лечение у этого доктора. В нашем случае со стороны эпилепсии ухудшений не было, и дочь уже после первого курса встала на ноги. Мы ездили к нему пять раз, каждые полгода. И после пятого курса она начала ходить. Криво — но ходить.
Потом мы начали закреплять этот эффект — с ней сейчас занимаются реабилитологи, Таисия встала на лыжи — с проектом «Лыжи мечты» Наташи Белоголовцевой. Она уже четвертый год катается на горных лыжах с тренером. Не боится, ей это нравится. А летом — на роликах.
— То есть прогнозы врачей не оправдались.
— Да. Возможно, еще и потому, что болезнь новая, мало изученная, и раньше такие дети не доживали до семи лет, а сейчас, при поддержке и реабилитации, качество и продолжительность жизни меняются.
— Сколько лет вашей дочери?
— 12, она подросток. Я думаю, что врачи, которые нас консультировали в Мюнхене, на тот момент еще не видели детей старшего возраста с таким диагнозом. Но я за последние годы познакомилась и со взрослыми людьми с мутацией CDKL5 — одной женщине 36 лет, другой 40. Конечно, многое зависит от родителей. Когда я стала искать международные организации, специализирующиеся на нашей болезни, то познакомилась с семьями из разных стран, с ассоциациями в Германии, Англии, США. И увидела, что дети и взрослые с нашей мутацией — очень разные. Есть такие, кто ходит, бегает, занимается спортом, как моя Таисия. Их мало. Есть такие, кто говорит «мама», «папа», их тоже мало, есть даже специальная международная группа в Facebook для детей с CDKL5, которые говорят. И в эту группу пускают только тех, чьи дети говорят. У нас в России тоже есть несколько таких детей, я их направила в эту группу, чтобы они были в сообществе. Это важно для родителей.
К сожалению, Таисия не говорит, она ментально не развита, она не узнает меня, не узнает отца. Такие дети живут в своем мире, они себя прячут очень глубоко. Но она чувствует прикосновения, тепло. Может реагировать на них, улыбаться.
Она может показывать, что ей нравится, а что — нет. Иногда что-то делает специально, то есть у нее характер проявляется. Но она не понимает меня, не понимает людей.
— Вы научились принимать ее болезнь?
— Я долго не принимала, потому что мне хотелось от ребенка отдачи, а ее не было. Нам трудно просто отдавать, ничего не получая взамен. Когда ей было пять лет, я поняла, что болезнь неизлечима. И приняла ее. И начала улучшать качество жизни дочери. Мы с ней ходим к логопеду, к психологу. Она произносит какие-то дополнительные звуки, но это не речь. Я купила ей аппарат, который управляется при помощи глаз, и человек может сказать «да-нет», «хочу—не хочу». Но Таисии этот аппарат не помог коммуницировать с миром. У меня появилась подруга из немецкой Ассоциации CDKL5, у нее дочь такого же возраста с такой же болезнью, она не ходит, но глазами через прибор сообщает о своих желаниях. Разные дети, и болезнь проявляется по-разному.
— До 2012 года мутация CDKL5 считалась одной из разновидностей синдрома Ретта, а потом для нее создали отдельный код в Международной классификации болезней. В чем различие между этими двумя болезнями?

Почему людей с тяжелыми нарушениями развития нельзя лишать права жить на воле
— Да, первоначально всех наших детей считали больными атипичным синдромом Ретта. Но в 2012-м мутацию CDKL5 выделили в отдельное заболевание. При синдроме Ретта ребенок развивается самым обычным образом, он даже может пойти и заговорить. Но с первым приступом эпилепсии начинаются задержка развития и резкая деградация. У детей с синдромом Ретта все время происходит откат назад, потеря навыков. У наших, с мутацией CDKL5, возможны хотя бы миллиметровые подвижки.
— Почему вы решили открыть ассоциацию в России, можно же было просто стать частью какой-то международной организации?
— В 2019 году я была в Риме на Всемирном форуме CDKL5-мутации. Я была там одна из России. И мне тогда итальянцы предложили: «Вам надо создать организацию в России». Понимаете, это же важно для государства — если у тебя есть такие дети, ты делаешь что-то для улучшения их жизни. Я говорю: «Да на это столько сил надо, у меня нет времени, я одна воспитываю дочь». А потом вернулась в Россию, и ко мне как-то вдруг стали семьи обращаться. То врач даст телефон мой, то в Facebook мне кто-то напишет.
Смотрю, уже пять, шесть, семь семей. Я поняла, что им нужны поддержка и помощь. Чаще всего — моральная поддержка. Это было началом пути. Я создала группу в Facebook, сейчас там около ста человек, и мамы, и папы. И группа в WhatsApp у нас есть, там 68 семей — только в этом году добавилось 10. Наша ассоциация пока не зарегистрирована, мы планируем это сделать.
У меня нет наполеоновских планов. Я просто хочу что-то сделать для таких детей, как моя дочь.
Мы не просим чего-то сверхъестественного. Мы хотим, чтобы о нас знали. Чтобы врачи в регионах знали, куда направить такого ребенка. Потому что его надо сразу отправлять на генетический анализ, который делается в НПЦ в Солнцево, в НИИ педиатрии и в некоторых частных лабораториях в Москве. Анализ этот — платный, 30 тыс. рублей. Часто у родителей нет таких денег, они ищут их через фонды. Кому-то помогают, кому-то нет.

Ассоциация родителей, воспитывающих детей с мутацией CDKL5, совсем молодая, в прошлом году ее участники встретились впервые в Москве
Фото: предоставлено Марией Ивановой-Сурае
Ассоциация родителей, воспитывающих детей с мутацией CDKL5, совсем молодая, в прошлом году ее участники встретились впервые в Москве
Фото: предоставлено Марией Ивановой-Сурае
— Диагностика в России за последние годы улучшилась?
— Да, я считаю, что заметно улучшилась. В Москве и в Петербурге уже делают анализ на нашу мутацию. Некоторые врачи из государственных научных медицинских центров знают про эту мутацию, генетики про нее знают. Выявив мутацию, генетики направляют наших детей к неврологам, потому что у каждого ребенка с таким диагнозом есть эпилепсия. Приступы — разные. У кого-то каждый день, у кого-то раз в месяц, у кого-то длительная ремиссия.
«Когда в Европе был локдаун, мы делились друг с другом лекарственными запасами»
— Чем лечат детей с мутацией в гене CDKL5?
— От мутации лекарства пока нет, но нашим детям необходимы препараты от эпилепсии. И большинство этих препаратов — не зарегистрированные в России. Нам приходится их доставать, часто подпольно. Помните, одна мать заказала из-за границы фризиум, и на нее дело завели? Он же тоже против эпилепсии. Вот наши родители каждый раз рискуют. Есть федеральный фонд «Круг добра», в который мы обратились с просьбой помочь нам с лекарствами, но они нас не приняли в фонд, потому что у нас мутация неизлечимая, а они принимают только тех, у кого заболевание лечится. Наша мутация — жизнеугрожающее, редкое, неизлечимое заболевание. Но и в нашем царстве слез есть луч надежды — это международный фонд LouLou, основанный семьей Джафар, в которой тоже родился ребенок с мутацией CDKL5. Это очень богатая ливанская семья — муж, Мажид Джафар, из Ирака, жена, Линн, — и они оплачивают все мировые исследования, в рамках которых ученые пытаются найти препарат против этой мутации, а также разрабатывают новые противоэпилептические препараты. 18 лабораторий мира работают за счет грантов этого фонда. Мы тоже с ними знакомы, они проводят международные научные форумы, ездят на них, оплачивают для семей, которые хотят попасть на эти форумы, проезд и проживание. Мы от нашей Ассоциации ездим туда бесплатно, они оплачивают билеты для главы ассоциации, врача, а, когда мы летали в Бостон, оплатили еще участие нашего переводчика и двух волонтеров. Мы получаем на этих форумах самую последнюю информацию, переводим ее и размещаем в группе, так что наши родители знают, какие исследования идут, на каком они этапе и так далее.
— И на каком этапе находятся эти исследования?
— Сейчас несколько лабораторий разработали препараты против этой мутации, один уже испытывают на животных.
Наши ученые-генетики тоже хотят участвовать в таких исследованиях, у них большой интерес, у нас мощная генетика, мировое сообщество готово давать гранты на такие исследования в России, но почему-то в нашей стране такие исследования не приветствуются.
Часто нам говорят, что у государственных учреждений нет валютного счета, и они просто не могут такие гранты принять.
— А на каком уровне вы сталкиваетесь с такими опасениями? На федеральном?
— Нет, скорее на уровне Московского департамента здравоохранения, они почему-то боятся делать совместные исследования, не хотят, чтобы наши генетики получали гранты, например, от американцев. Хотя Пенсильванский университет и фонд LouLou дают ежегодные гранты на такие исследования. А ведь было бы здорово работать над этим вместе.
— Теми препаратами, которые уже создаются, можно будет вылечить детей с мутацией гена CDKL5?

Основатель благотворительного фонда «Семьи СМА» Ольга Германенко о жизни пациентов
— Думаю, у маленьких детей точно есть шансы. Я ничего не могу сказать про наших подростков или про взрослых. Скорее всего, лекарство будет эффективным для малышей, как и в целом любые препараты генной инженерии. Но все наши семьи живут надеждой.
Раньше, кстати, эта мутация касалась только девочек: мальчики умирали сразу после рождения. Но потом они стали выживать, они лежат, у них нарушения сильнее, чем у девочек, но они живут. А следующее поколение мальчиков — другое, некоторые уже сидят и ходят. Так что эта мутация тоже подвержена каким-то изменениям, как будто приспосабливается к человеку.
— Это не новая мутация, она была всегда?
— Трудно сказать, но, скорее всего, да, она была и раньше. Просто ее не диагностировали. Раньше детям с младенческой эпилепсией ставили диагноз «эпилепсия» или ДЦП и не выясняли ее причины. Сейчас в мире стараются выявить причины, которые приводят к эпилепсии. И одна из таких причин — мутация CDKL5. Пока не знаешь причину, невозможно лечить эпилепсию. Бывает эпилепсия, связанная с родовой травмой, и ее можно вылечить. А бывает эпилепсия, вызванная генетическими причинами,— и тут важно принять, что она есть и будет, но ты можешь работать над частотой и продолжительностью приступов, над их предупреждением. Потому что каждый приступ опасен. Ребенок в этот момент не дышит, теряет сознание, клетки мозга отмирают. Ребенок может погибнуть. И такие приступы тормозят его развитие.
У наших детей эпилепсия не такая, как в кино показывают, когда человек падает с пеной у рта. У нас эти приступы выглядят иначе. Ребенок замирает, у него каменеют ноги-руки, он как будто смотрит в одну точку, но он без сознания, ничего не видит, не слышит.
И все, что ты можешь,— быть с ребенком рядом, гладить его, разговаривать. Приступ может длиться минуту, может — пять минут. Потом ребенок сам из этого состояния выходит, начинает дышать. Но каждый такой приступ может стать последним. Бывает и так, что наступает непрерывный эпистатус, ребенок не выходит из приступа. Тогда его кладут в реанимацию. Дети с нашей мутацией там умирают быстрее. Поэтому очень важно купировать приступы и не допускать попадания в реанимацию.
— То есть родитель не просто должен ждать, пока приступ пройдет, а давать ребенку препараты?
— Как вы покупали эти препараты во время пандемии?
— Нам повезло, что у нас есть сообщество. Наши семьи — это очень позитивные люди, добрые, открытые. И мы друг другу помогаем. Когда в Европе был локдаун, группа наскребала по своим сусекам для тех детей, у кого запас закончился. Мы делились друг с другом лекарственными запасами. Я искала диазепамовые свечи, которые не зарегистрированы в России,— они быстро снимают эпилептический приступ. Я бы хотела, чтобы Таисия сама умела выходить из этого состояния. Но на всякий случай я хотела приобрести эти свечи, чтобы они лежали у меня дома, и я была бы спокойна: вдруг у дочери приступ перейдет в эпистатус, а скорая не приедет быстро, и тогда эти свечи помогут. Родители готовы пойти на все ради того, чтобы иметь такие лекарства. Мне одна мама из Подмосковья отправила это средство.
Я считаю, что наше государство все-таки должно об этом позаботиться. Да, многие говорят: процесс пошел. Ну он уже полтора года идет, этот процесс. И ни сабрил пока не зарегистрирован, ни букколам, ни фризиум, ни диазепамовые свечи. Мы все еще ждем.

Дети с мутацией гена CDKL5 нуждаются в лекарствах, которые не зарегистрированы в России. Это держит их родителей в напряжении и заставляет искать способы получения препаратов
Фото: Архив Марии Ивановой-Сурае
Дети с мутацией гена CDKL5 нуждаются в лекарствах, которые не зарегистрированы в России. Это держит их родителей в напряжении и заставляет искать способы получения препаратов
Фото: Архив Марии Ивановой-Сурае
— В России вообще нет зарегистрированных препаратов, которые могли бы снижать частотность эпилептических приступов?

Как корреспондент “Ъ” провела день с подопечным детского хосписа
— Есть препарат ганаксалон — его исследовали в НИКИ педиатрии, он показал снижение количества приступов на 30%. И мы ждем, что в этом году он уже появится на российском рынке. Пока неизвестно, сколько он будет стоить, как его будут выписывать. Я надеюсь, что процесс закупки лекарств, выдачи льготных рецептов будет не сложным для родителей. Сейчас выбить какой-то рецепт — это целый подвиг. Я уже не говорю про отношение: ты приходишь в поликлинику, в соцзащиту — и как будто сразу всем там что-то должен. Ни о каком дружелюбии речи не идет. А хотелось бы прийти туда, где тебе помогут, где тебя защитят и подскажут, как правильно действовать.
— Вы сказали, что у вас в ассоциации 68 семей, воспитывающих детей с мутацией CDKL5. А в России их сколько?
— Тысячи. 68 — это число выявленных, диагностированных детей, живущих в крупных городах, которые нашли друг друга. В мире их — миллионы. Если один случай на 40 тысяч, посчитайте, сколько их может быть в России. И число детей с таким диагнозом растет по всему миру.
— Какой самый взрослый ребенок с такой мутацией живет сейчас в России?
— У нас в ассоциации такому ребенку 16 лет. А самому маленькому — полгода. Многие семьи, кстати, рожают других, здоровых, детей. Но женщины с трудом на это решаются, им нужен психолог. Мы сами в своем сообществе поддерживаем этих женщин, они испытывают огромный страх.
— А пренатальный скрининг не делают?
— Делают, и на эту мутацию нам тоже предлагают сделать. Но не все готовы на такое вмешательство, это же травматичная процедура.
«Департаменты соцзащиты работают на то, чтобы никого не защищать»
— Как вам кажется, в обществе отношение к детям и взрослым с инвалидностью меняется?
— В прошлом году мы провели первую встречу семей, воспитывающих детей с мутацией CDKL5 в России. Семьи из 28 городов приехали в Москву, с билетами и гостиницей нам помог как раз фонд LouLou. Многие приехали с другими детьми, с отцами. Мне очень понравилось, что у многих полные семьи, отцы их не бросают, помогают и дают своим женам отдохнуть. И в этом году, 1 июня, мы готовим вторую такую встречу, на нее приедет 145 человек.
Но было очень сложно найти отель для нашего мероприятия. Единственный отель, который согласился принять наши семьи,— Holiday Inn на Павелецкой. Мы отправили порядка 50 писем в разные отели, но нам отвечали: «У нас нет пандусов», «Мы переполнены». На самом деле причина, конечно, не в этом. Просто общество пока не готово видеть детей в тяжелом состоянии, в инвалидных колясках.

Как лечат в России пациентов с редкими болезнями
Мы с дочерью ходили в бассейн на Можайском шоссе, ей дали время — 10 утра в воскресенье. Как-то она пошла с няней без меня, они опоздали, и их не пустили, потому что там уже были другие люди, а в ее время в бассейне никого нет. И это в Москве, где в целом еще более или менее можно жить. Мы, например, с дочерью и в кафе ходим, и в рестораны, я ее не скрываю, и я не вижу в общественных местах негативного отношения. Может быть, что-то стало меняться, после того как Наталья Водянова стала рассказывать о своей сестре, а другие известные люди — о своих детях. Я вижу, что отношение меняется, но очень медленно.
В регионах ситуация хуже, отношение к нашим семьям, особенно со стороны чиновников соцзащиты, плохое.
Мы постоянно боремся за путевки в санатории для наших семей. Эпилепсия считается противопоказанием для реабилитации. Таисия у меня занимается горными лыжами, иногда выигрывает медали. Я считаю, что, например, реабилитация в паралимпийском центре «Эволюция» в Крыму ей показана. Но мы ни разу туда не попадали. Вот в этом году нам сказали: «Возможно, вы поедете в июне, но решим мы это в мае». О том, что мы планируем лето заранее, никто не думает. И дадут нам эту путевку или нет — будет решать комиссия, которая состоит не из врачей. Она состоит из чиновников. Почему чиновник, заместитель главы округа, должен принимать решение, показана моей дочери реабилитация в этом центре или нет?
По закону дети с инвалидностью имеют право на две путевки на реабилитацию в год, но об этом семье никто не скажет. Если дали одну путевку — скажи «спасибо».
Некоторые наши родители пишут, что их дети не были на реабилитации ни разу. В санатории — ни разу. Потому что они не борются за свои права. Они по десять лет сидят дома, никуда не выезжают, они очень скромные, и им никто ничего не подсказывает. Хотя им положено каждый год выезжать на отдых и два раза в год — на реабилитацию. А чиновники от соцзащиты молчат, как будто свой карман берегут. Многие люди годами стоят в очереди на путевки в санатории и не получают такой возможности.
— То есть система соцзащиты работает плохо.
— Да. Я вообще считаю, что департаменты соцзащиты свое называние не оправдывают абсолютно. Они работают на то, чтобы никого не защищать. Многим семьям трудно даже памперсы получить, которые им обязаны выдавать, потому что они прописаны в ИПР (индивидуальная программа реабилитации.— “Ъ” ). То размер не тот дадут, то количество. Говоришь: «Ребенок вырос, ей мало четырех памперсов в день», а тебе отвечают: «У нас максимум — пять». Вот эти «максимум пять» памперсов рассчитаны на человека от года до 18 лет. То есть никак не учитывается, что дети растут. Или, например, у нас в ИПР записан жесткий корсет, а ортопед прописал дочери мягкий. И, чтобы получить мягкий корсет за госсчет, мне надо снова идти с ребенком на врачебную комиссию и вносить изменения в ИПР. А комиссия занимает кучу времени. И, даже если ты записываешься на определенное время, ты все равно стоишь там несколько часов. А с нашими детьми это очень трудно. То есть вся система создана не для того, чтобы упрощать нашу жизнь, а для того, чтобы делать ее сложнее.
Я придумала проект «Вишневый город» — это такой мини-город, где есть отель, ресторан, апартаменты, научный медицинский центр, реабилитационный центр для детей с инвалидностью, кинотеатр для всех, бассейн для всех, общая школа, общий детский сад, университет, мастерские, сад ощущений. То есть такое инклюзивное место, где комфортно всем. Ведь нашим детям практически некуда пойти. У них нет реабилитации, нет занятости, даже в Москве нет полноценных мастерских, чтобы все было в одном месте, куда можно было бы интегрировать самых разных детей с инвалидностью. В Пскове есть, а в Москве нет. Я нашла архитектора, мне помогли сделать презентацию.
Мы хотели, чтобы этот «Вишневый город» был где-то в центре, в шаговой доступности от метро. Я отправила презентацию мэру Москвы, он спустил ниже, в департамент соцзащиты. Меня пригласили к какому-то чиновнику среднего уровня, и она, даже не просмотрев эту презентацию, спрашивает: «Вы уверены, что этот проект городу нужен?» Ну что я могу ответить на этот вопрос? Вместо того чтобы провести мониторинг проблем семей, воспитывающих детей с инвалидностью, изучить статистику, выяснить, сколько детей и взрослых с инвалидностью сидят запертыми дома, эти чиновники задают мне риторические вопросы. В общем, диалог не получился. Потом мне сказали: «Ты идешь снизу вверх, у тебя ничего не получится. Надо идти сверху вниз». То есть, кроме «Прямой линии» с президентом, других путей нет.
«Когда-то я поставила цель — улучшить качество ее жизни, к этому я и иду»
— Вы работаете, при этом одна воспитываете ребенка. Как вам это удается?
— С моей дочерью находятся две няни. Я сейчас много работаю, у меня своя кинокомпания, недавно состоялся мой режиссерский дебют в игровом кино, три месяца шли съемки в Ливане. Конечно, без нянь я не справилась бы.
— Сложно найти няню для детей с такими особенностями?
— Одну няню я нашла через частный ресурс, она работала с детьми с аутизмом в Саратове, и она у меня уже три года. Вторая няня с Украины по профессии повар — отличная, добрая женщина. Мне пришлось самой их обучить, показывать видео, что такое эпилепсия, что делать в случае приступа.
— Они могут дать Таисии лекарство, если случится приступ?
— Могут, но сейчас нужно просто обнять ее и ждать, пока приступ пройдет. Таисия сама умеет выходить из этого состояния. Но на крайний случай есть инструкции, которые няни знают.
— Вы сказали, что ваша дочь не понимает обращенную к ней речь. А как проходит обучение Таисии?

Как в России живут дети с неизлечимыми заболеваниями
— На уровне рефлексов. С тренером она занимается три года по два раза в неделю, тренер держит ее за руку, ставит ей ноги, так она научилась кататься на лыжах. С нашими детьми хорошо работает поведенческая терапия. Например, Таисия любит сыр. Ее приучают — ты получишь сыр, когда возьмешь сама ложку. Вот такие занятия у нее уже несколько лет, она умеет и ложку держать, есть сама, пить из кружки. Может стол вытереть, ее научили. Но она не может себя обслужить.
— Педагоги приходят к вам домой?
— Да, у нас семь педагогов. И три раза в неделю Таисия ездит в специальную школу, она находится там с 10 до 15 часов, у нее в школе ЛФК, бассейн, психолог, логопед, музыка, арт-терапия. Там есть сенсорные комнаты, групповые занятия. Я очень рада, что она как-то взаимодействует там с другими детьми,— может мальчика за руку взять. Она там не одна. Когда-то я поставила цель — улучшить качество ее жизни, к этому я и иду. Это все очень дорого стоит, мне приходится очень много работать. Но я стараюсь. И я считаю, мне еще повезло — у меня есть возможность обеспечить своего ребенка необходимым. У многих родителей этой возможности нет.
— Чего еще помимо реабилитации и зарегистрированных лекарств не хватает семьям, воспитывающим детей с таким диагнозом?
— Родителям не хватает бесплатного психолога — и мамам, и папам. Многие родители хотели бы работать, но у них нет возможности нанять няню, да и нянь надо готовить, не каждому человеку можно доверить ребенка с тяжелыми нарушениями. Вот мы хотим, чтобы в Москве и в регионах были проекты, которые готовили бы нянь и предоставляли бы их семьям бесплатно. В Москве есть такие частные проекты, а я считаю, что должен быть государственный проект, потому что это большие затраты. Очень нужна вот такая точечная помощь семьям, у которых тяжелые дети. Есть одинокие мамы, воспитывающие сложных детей, они не могут работать, не могут нанять нянь, они в тяжелом положении. Если дать такой маме няню, она выйдет на работу. А кому-то надо помочь найти работу, и это тоже задача государства, которое должно защищать всех своих граждан.