Синдром денди уокера прогноз для жизни
Главным признаком классической мальформации Денди-Уокера является полная или частичная агенезия червя и крупная кистозная мальформация в задней ямке, соответствующая дивертикулярному расширению крайне увеличенного четвертого желудочка. Гидроцефалия часто отсутствует у новорожденных и детей раннего возраста и иногда до позднего периода полового созревания. Обычно имеется заметное расширение задней ямки и возвышение места слияния синусов и боковых синусов, проходящих по теменным костям вместо чешуи затылочной кости.
В соотвествии с недавним определением, используемым и в статьях на сайте, сочетанные соматические мальформации не характерны. Сопутствующие пороки развития ЦНС включают патологическую миграцию в нижней оливе, агенезию мозолистого тела у 7-15% пациентов (Hirschetal, 1984, Murray et al., 1985) и затылочные цефалоцелеу 17% (Hirsch et al., 1984). Периферические мальформации присутствуют в широко изменяющейся доле случаев, в зависимости от используемых диагностических критериев и от происхождения случаев. В ряде диагностированных пренатально случаев, частота сопровождающих мальформаций может быть высокой. Сюда входят периферические мальформации, такие как расщелина губы и неба, пороки развития сердца, аномалии мочевыделительного тракта и малый лицевой дисморфизм (Has et al., 2004).
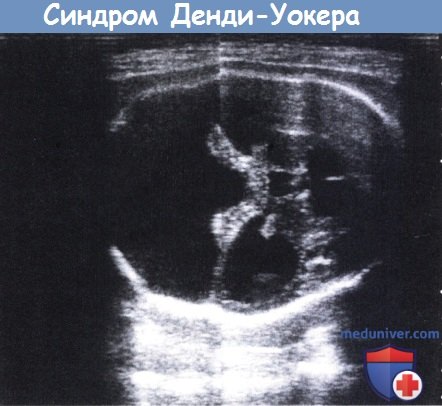 Мальформация Денди-Уокера.
Мальформация Денди-Уокера.
Пренатальная диагностика методом ультразвукового сканирования на 33 неделе беременности.
Хорошо видно отсутствие червя мозжечка и выраженное расширение задней черепной ямки (левая часть рисунка).
Синдром Денди-Уокера, в строгом понимании, является только редким семейным состоянием, хотя имеются редкие сообщения о развитии этого состояния у сиблингов. В подавляющем большинстве случаи спорадические. Хромосомные аномалии, включая частичную делецию хромосомы 13q, встречались в нескольких случаях (Murray et al., 1985, Bordarier и Aicardi, 1990, Ballarati et al., 2007). Недавно в нескольких случаях синдрома Денди-Уокера обнаружили промежуточную делецию в хромосоме 3q2, охватывающую гены ZIC2 и ZIC4, как считают, отвечающую за мальформацию (Grinberg et al, 2004).
Основным клиническим проявлением синдрома Денди-Уокера является гидроцефалия. Поскольку гидроцефалия обычно становится заметной к концу первого года жизни, в 75% случаев диагноз ставится в возрасте после трех месяцев и редко в первый день рождения. Голова может быть вытянутой в переднезаднем направлении с выступающим затылком, что может натолкнуть на мысль о возможном наличии аномалии. Примечательно, что отсутствуют мозжечковые симптомы или нарушения позы. Некоторая степень задержки умственного развития отмечается в 30-50% случаев, что является показанием для проведения нейровизуализации.
Визуализация имеет характерную картину. Во всех случаях присутствуют огромные скопления, которые в половине случаев распространяются через тенториальное отверстие в тригеминальную цистерну и/или вниз через большое затылочное отверстие. Это проявляется значительным расширением четвертого желудочка. Полушария маленькие, но нормальные. Червь мозжечка часто уменьшен и смещен, прижат к намету. Сагиттальные срезы позволяют лучше увидеть остатки червя, которые смещены вверх к намету мозжечка и повернуты кпереди. По мнению Klein et al. (2003), у большинства пациентов имеется только две щели червя и три доли; такие пациенты не имеют сочетающихся мальформаций и их уровень развития почти всегда остается в пределах нормы. В наименьшей группе червь развит неправильно и имеет только одну щель или ни одной. В этих случаях всегда присутствуют сопутствующие аномалии и большая или меньшая задержка умственного развития.
Дифференциальная диагностика мальформации проста в типичных случаях. В мегацистерне большой ретроцеребеллярный карман (соответствующий карману Блейка) сообщается с четвертым желудочком, но червь мозжечка полный. Дифференцировка варианта синдрома Денди-Уокера от увеличенной цистерны магна или ретроцеребеллярной арахноидальной кисты представляет некоторые трудности (Tortori-Donati et al., 1996, Boltshauser et al, 2002). В вариантных формах червь может быть атрофичным, но содержит все дольки и сдавлен стволом. В случаях с мегацистерной червь по сути нормальный, и четвертый желудочек отличается от кисты. В случаях Денди-Уокера червь может быть аномальным, атрофированным, неполным или вообще отсутствовать, четвертый желудочек трудно отличить от кисты и переднее положение сосудистого сплетения смещено к верхней стенке кисты, что является значимым диагностическим признаком (Nelson et al., 2004). Намет мозжечка смещен вверх, его вырезка видна на теменной, а не на затылочной кости, приводя к значительному расширению задней ямки.
Пренатальная диагностика относительно проста с 18-й по 20-ю неделю.
Исход для пациентов, страдающих синдромом Денди-Уокера, относительно неблагоприятный при использовании обычных критериев. Уровень смертности в 144 случаях, рассмотренных Hirsch et al. (1984), составил 27% и только около половины выживших имели нормальный IQ. Напротив, исход благоприятный при применении строгих параметров Klein et al. (2003), если оперативное вмешательство по поводу гидроцефалии выполнено рано (Parisi и Dobyns, 2003). Генетический прогноз хороший, с рецидивами в 1-5% случаев. Это контрастирует с более тяжелым прогнозом атипичных и/или ассоциированных форм. Хотя увеличенная цистерна магна обычно рассматривается как доброкачественное изменение, Bodensteiner et al. (1988) обнаружили, что у 62% пациентов с мегацистерной магна была неврологическая патология, полагая, что это может быть частью комплексной мальформации. Случаи Денди-Уокера-подобного синдрома задней ямки, связанные с лицевыми или другими гемангиомами и сердечными и сосудистыми аномалиями, представляют различные формы (PHACE/PHACES синдромы или синдром Паскаля-Кастровьеджо II) (Metry et al., 2001).
Лечение заключается в шунтировании гидроцефалии, а не в открытии кистозного четвертого желудочка. Некоторые хирурги советуют устанавливать шунт в четвертый желудочек, но это мнение не единодушное. После шунтирования боковых желудочков может потребоваться дополнительное шунтирование четвертого желудочка. Хорошие результаты может дать эндоскопическая хирургия (Mohanty et al., 2006).
Механизмом синдрома Денди-Уокера ранее предполагалась атрезия отверстий Мажанди и Люшки. Тем не менее, эта атрезия не является ни постоянным, ни характерным признаком. В настоящее время этот синдром связывают с остановкой развития среднего мозга с сохранением переднего мембранозного участка четвертого желудочка плода. Эта структура в норме исчезает до момента открытия отверстия. Аномалия, по всей видимости, появляется до третьего месяца беременности (Friede, 1989). Полная атрезия отверстия четвертого желудочка редко наблюдается вне комплекса Денди-Уокера (Amacher и Page, 1971). Это вызывает формирование гидроцефалии, которая у одного из моих пациентов началась и была более заметна в четвертом желудочке, чем в остальных частях желудочковой системы. Микроскопическое исследование оболочки, покрывающей четвертый желудочек, выявило глиальную ткань, выстланную по внутренней стороне эпендимой (Friede, 1989). Эта глиальная оболочка позволяет отличить атрезию отверстия от фиброза базальной цистерны с последующей гидроцефалией. Происхождение мембраны неизвестно.
Редактор: Искандер Милевски. Дата публикации: 30.11.2018
Аномалия Денди-Уокера (клинический случай)
Войнова Л.В., Чумакова Г.Н.
Областная детская клиническая больница, Северный государственный медицинский университет, г. Архангельск.
Этиология синдрома неизвестна. СДУ может быть проявлением генетических заболеваний, таких как синдром Меккеля (аутосомно-рецессивный путь- микроцефалия, полидактилия, поликистоз почек, глазные аномалии – микрофтальм, гипоплазия ЗН, ВПС, крипторхизм, затылочная спино-мозговая грыжа), Меккеля-Грубера (то же + энцефалоцеле), Варбурга, Тернера, трисомия 9, триплоидия, 6р- и другие хромосомные аберрации. Предполагается, что определенную роль играют и такие факторы как вирусная инфекция (ЦМВ, краснуха), алкоголь, диабет беременной.
Кроме пороков ЦНС часто встречаются ВПС, энцефалоцеле, поликистозные почки, полисиндактилия, гемангиомы, расщепления губы/неба, с Клиппеля-Фейля и др.
Впервые синдром описан Денди и Блекфаном в начале 20 века, они считали это состояние вторичным по отношению к врожденной атрезии отверстий Мажанди и Люшки, обспечивающих отток ликвора из 4 желудочка в субарахноидальное пространство; Уолкер был врачом, впервые проведшим хирургическое лечение больному с этой патологией, поэтому заболевание стало называться СДУ.
При УЗИ особое внимание нижней части мозжечка, хотя для окончательного диагноза все же необходимо проведение ЯМРТ мозга. Но при ЯМРТ возможны трудности при интерпретации результатов, поэтому идет поиск наиболее информативных методов диагностики- некоторые авторы предлагают радиоизотопные методы исследования, другие – использовать пневмоэнцефалографию. Обязательно кариотипирование для исключения хромосомной патологии. Основное проявление – прогрессирующая гидроцефалия при закрытой форме синдрома СДУ.
В отделении патологии новорожденных АОДКБ в 2006 году находился ребенок, у которого СДУ был заподозрен антенатально и подтвержден после рождения. Приводим описание данного случая.
При рождении: масса 3430 г., рост 50 см, окружность головы 37 см., окружность грудной клетки 35 см., оценка по шкале Апгар 8/9баллов. Период ранней адаптации: пастозность тканей, склонность к гипотермии, негрубый систолический шум, кривошея, тремор подбородка, синдактилия.
Статус при поступлении: состояние средней тяжести, беспокоен, грубый голос, непостоянное сходящееся косоглазие, брахицефалическая головка, БР 2,5-3см, установка головки и тела вправо, тремор кистей, спонтанный рефлекс Моро, мышечный тонус повышен по пирамидному типу, больше справа, СХР оживлены. Кожа розовая, чистая. Выделения из глаз, больше справа. Синдактилия 2-3 пальцев обеих стоп. В легких – без патологии, ЧД 38 в мин., ЧСС 144 в мин., негрубый систолический шум по левому краю грудины.
Заключение: вариант СДУ. Умеренно выраженная смешанная гидроцефалия. Нейрохирург – показаний для оперативного лечения нет. Осмотр окулиста от 25.07.06 – опалесценция роговицы, сосуды на глазном дне сужены, наблюдения в плане врожденной глаукомы, от 1.08.06 – опалесценция роговицы уменьшилась.
Проведено лечение: 10% глицерин, пантогам, местное лечение конъюнктивита. Состояние при выписке удовлетворительное.Вес4255(+825), окружность головы 38(+1см).. Выписан домой с рекомендациями – наблюдение у невролога, контроль ОГ 1 раз в 7 дней- 1 мес, затем по состоянию, НСГ через 1 мес, осмотр окулиста через 7 дней- (врожденная глаукома?), наблюдение кардиолога. ЗРТ+ 11.07, 13.07, 17.07.
Таким образом, ребенок с выявленным антенатально и подтвержденным после рождения пороком головного мозга (СДУ) в сочетании с пороком сердца, синдактилией и признаками текущей внутриутробной инфекции требовал ежемесячного катамнестического мультидисциплинарного наблюдения в условиях клиники для своевременного выявления и коррекции нарушения функции поврежденных органов и систем.
Литература.
Под редакцией академика РАМН Вельтищева Ю.Е., профессора Темина П.А.. «Наследственные болезни нервной системы» Москва. «Медицина» 1998г
Бадалян Л.О «Детская неврология» Москва « Медицина»,1984г.
Э.Парайц-Й.Сенаши «Неврологические и нейрохирургические исследования в грудном и детском возрасте» Издательство академии наук Венгрии,Будапешт 1980г.
Барашнев Ю.И., Бахарев В.А., Новиков П.В. «Диагностика и лечение врожденных и наследственных заболеваний у детей» Триада-Х, Москва, 2004г.
Под редакцией Кулакова В.И., Барашнева Ю.И. «Новорожденные высокого риска» издательская группа «ГОЭТАР-Медия», 2006 г.
Синдром Денди-Уокера — проявления и лечение

Среди врожденных пороков выделяют синдром Денди-Уокера. Причины болезни продолжают выяснять. Отклонение в развитии плода диагностируют методом УЗИ. Редко дефект мозга незначителен и качество жизни ребенка не ухудшается. У 50 % детей с этой аномалией сразу устанавливают инвалидность, минимум 25% новорожденных умирают. Возможности медицины позволяют устранить симптомы порока и облегчить состояние малыша. Лечение должно быть комплексным.
Что это такое?
Синдром Денди-Уокера принадлежит к группе неизлечимых врожденных пороков, влияющих на функции нервной системы. В справочнике МКБ―10 болезни присвоен код Q03.1: «Атрезия отверстий Мажанди, Лушки».
Впервые дефект описал в 1921 г. нейрохирург Уолтер Эдвард Денди. Он изучал причины гидроцефалии. Независимо друг от друга, исследованием также занимался Артур Эрл Уокер. В 1942 г. издали его статью об отсутствии протоков Лушки, Мажанди. Работы нейрохирургов взяты в основу изучения порока.
Особенности синдрома Денди-Уокера:
Синдрому Денди-Уокера характерна аномалия строения мозжечка и гидроцефалия. Четвертый желудочек мозга расширяется. Параллельно в зоне задней черепной ямки образуется киста с ликвором, поскольку пути циркуляции (отверстия Лушки, Мажанди) сужены либо отсутствуют. Также выделяют аномалию червя: соединяющая структура долей мозжечка не развивается или неправильно формируется.
Причины и триггеры у детей
Экспертами доказательной медицины не установлено точных причин врожденной аномалии мозжечка. Врачи считают, что синдром Денди-Уокера возникает из-за генетического нарушения, хромосомного расстройства, чрезмерной продукции церебральной жидкости. К триггерам (провоцирующим факторам) относят влияние токсических и вредных веществ, отравляющих организм женщины.
Справка! Риск врожденного порока у плода при повторной беременности не превышает 7%, если нет генетической предпосылки у матери или отца.
Риск порока у плода повышается у беременных женщин:
Генетическое расстройство может спровоцировать радиация или цитомегаловирус, краснуха, иные вирусные инфекции во время беременности. Триггером также считают наличие у женщины сахарного диабета, аутоиммунной болезни, метаболического нарушения. Большую роль играет наследственность, то есть, в семейном анамнезе уже регистрировались врожденные пороки.
Симптомы и признаки
До рождения младенца беременная женщина не ощущает какого-либо нарушения или осложнения гестации. У плода на УЗИ выявляют только пороки мозжечка: недоразвитие, увеличение четвертого желудочка, образование ликворной кисты.
После рождения к ультразвуковым признакам синдрома Денди-Уокера добавляется неестественное поведение младенца. Из-за головной боли он постоянно плачет. Сдавливание мозга кистой приводит к судорогам, подергиваниям конечностей, повышению мышечного тонуса. Это проявляется нарушенной моторикой, непроизвольным вращением глаз, суставы рук и ног согнуты.
К признакам врожденного дефекта мозжечка и ликворных путей относят:
По мере взросления проявляется умственная отсталость, нарушение двигательных навыков (координации). Ребенок капризен, раздражителен, не может ровно ходить, устойчиво стоять. Периодически случаются приступы судороги. По утрам возникает рвота, тошнота, усиливается головная боль. При обследовании выявляют ухудшение зрения.
У 50% детей выраженных симптомов нет. У ребенка из этой группы голова не деформирована. Но координация движений нарушена, походка качающаяся, ноги во время ходьбы широко расставлены. Есть незначительное отставание в развитии психомоторики, умственной деятельности.
Справка! Редко симптомы и признаки врожденного дефекта проявляются у детей после 4 лет или у взрослых людей.
Синдром Денди-Уокера у взрослых
В статистике есть сведения, когда дефект мозжечка диагностировали после совершеннолетия человека. Нарушение чаще относят к разновидности водянки мозга. В этих случаях признаки и симптомы синдрома Денди-Уокера проявляются постепенно после 18 лет.
По мере ухудшения состояния:
К интеллектуальным нарушениям относят ухудшение памяти, потерю приобретенных навыков. Человек перестает узнавать родных и знакомых, становится раздражительным, разучивается читать, одеваться, писать и так далее.
Формы
В дородовой период врачи классифицируют полную и неполную формы порока. В первом случае мозжечок неразвит: отсутствует червь, киста соединяется с четвертым желудочком. При втором виде дефект образуется частично. Структура между долями на УЗИ местами просматривается, жидкостная полость сообщается не всей поверхностью с червем.
После родов врачи выделяют открытую и закрытую форму порока. Различие этих видов основано на проходимости ликворных путей. В первом случае есть сообщение желудочка через отверстия Лушки и Мажанди с подпаутинным пространством. При закрытой форме протоки заращены.
Диагностика
Женщину обследуют методом УЗИ раз в триместр. Ультразвуковое сканирование безвредно для плода, позволяет обнаружить аномалии на ранних сроках. Исследование мозга может выявить отклонения развития на 2,5 месяце беременности. Но для подтверждения синдрома Денди-Уокера повторно УЗИ делают на 20―23 неделе гестации. На этом сроке уже видно ликворную кисту, неправильное строение мозжечка, деформацию IV желудочка.
При повторном выявлении признаков женщине назначают МРТ. По форме врожденного порока врачи оценивают тяжесть синдрома Денди-Уокера. Ввиду неизлечимости болезни рекомендуют прерывание беременности. Но окончательное решение принимают родители ребенка.
После родов врач осматривает младенца, ощупывает и измеряет голову. Доктора оценивают симптомы, внешние проявления болезни, затем ребенка направляют на УЗИ внутренних органов, нейросонографию. У матери доктор выясняет анамнез семьи, образ жизни, течение беременности, случаи самолечения и вид употребляемых препаратов. Маме желательно проконсультироваться у генетика.
Важно! Неопытность узиста или доктора увеличивает риск ошибки. Для подтверждения диагноза беременной женщине надо проконсультироваться и/или дообследоваться у 2―4 врачей независимых медицинских учреждений.
Методы лечения
Пациентам с врожденным пороком и их родителям предоставляют паллиативную помощь. Сюда входят методы поддерживающей терапии, консультации психологов, социальных работников.
Медикаментозное и хирургическое лечение применяют при выраженных симптомах порока. Если нет внутричерепной гипертензии, гидроцефалии и прочих нарушений функций внутренних органов – ребенок находится под наблюдением врачей. Его должен регулярно обследовать нейрохирург, невропатолог, педиатр.
Детям с синдромом Денди-Уокера проводят поддерживающую терапию витаминно-минеральными комплексами, средствами аминокислот. Назначают также лекарства, устраняющие выраженные симптомы порока. Для восстановления работы сердца, почек, других органов препараты подбирают индивидуально по медицинским показаниям.
Препараты, назначаемые при синдроме Денди-Уокера:
Также назначается физиотерапия. В нее входят ЛФК, бальнеотерапия — лечебные грязи, плаванье, солевые ванны. Также врач назначает массаж. Эти методы направлены на снижение мышечного тонуса пациента, а также улучшение его психоэмоционального состояния.
Операция показана для восстановления оттока ликвора другим путем. Используют два метода: вентрикуло-перитонеальное шунтирование или эндоскопическую вентрикулостомию. В первом случае мягкую трубку вставляют в желудочек. Затем катетер подкожно за ухом и в области ключицы направляют в брюшную полость. В нее будет стекать и рассасываться жидкость.
При вентрикулостомии в черепной кости делают отверстие, продвигают в желудочек нейроэндоскоп, отводят лишний ликвор. Процедура малоинвазивная, недлительная, пациента выписывают на 3 день, поздних осложнений не фиксировалось.
Справка! Хирургическое лечение устраняет только гидроцефалию и внутричерепное давление. Умственные способности, координация движений или функции ЦНС операцией не восстанавливается.
Возможные осложнения
К осложнениям синдрома Денди-Уокера относят водянку, некроз мозга, дисфункцию внутренних органов, внутричерепное кровоизлияние. Вследствие порока нарушается зрение, работа центральной нервной системы, возникает психическая, интеллектуальная неполноценность. У старших детей пропадают уже приобретенные навыки. У 10% прооперированных шунты забивались, регистрировались случаи инфицирования.
Прогноз на жизнь
Для пациентов с синдромом Денди-Уокера нужен пожизненный уход, повышенное внимание. У детей прогноз жизни неблагоприятен. Инвалидность устанавливают сразу. Интеллект не поддается коррекции. Дети с тяжелым пороком требуют беспрерывной симптоматической терапии. Их продолжительность жизни катастрофически мала — они умирают в возрасте младше 3 месяцев. Люди с неполной формой аномалии живут долго. Благодаря лечению мышечный тонус и координация движений нередко нормализуется.
Профилактика
Специфической профилактики нет. Женщине надо вести здоровый образ жизни, избегать контактов с вредными веществами. В период планирования беременности нужно повторно обследоваться, вылечить выявленные половые инфекции. Во время вынашивания нельзя принимать лекарства с тератогенным или эмбриотоксическим свойством. Эти препараты также становятся причиной врожденных уродств и пороков.
В целях профилактики нельзя пропускать плановые посещения гинеколога, УЗИ. Раннее выявление инфекционно-воспалительных болезней и их лечение снижает риск врожденных пороков.
Медицинские интернет-конференции
Языки
НАСЛЕДСТВЕННАЯ ГИДРОЦЕФАЛИЯ (СИНДРОМ ДЕНДИ-УОКЕРА) КЛИНИЧЕСКИЙ СЛУЧАЙ
Лысова Ю.В.
Научный руководитель: к.м.н., доцент Нечаев В.Н.
Резюме
Количество врожденных пороков развития в последнее десятилетие заметно увеличилось. Пороки развития нервной системы суммарно занимают третье место в структуре аномалий развития после врожденной патологии сердечно-сосудистой и мочевыводящей систем, причем около 80% этих заболеваний представлены гидроцефалией различного генеза [1]. Среди большого количества возможных аномалий одним из наиболее тяжелых по своим последствиям считается синдром Денди Уокера.
Ключевые слова
Статья
Данный синдром был впервые описан американским нейрохирургом Уолтером Денди в 1921 году и Эрлом Уокером в 1944 г. Среди живорождённых детей частота встречаемости от 1:5000 до 1:25000, а среди детей с врождённой гидроцефалией колеблется от 3,5 до 12% [2].
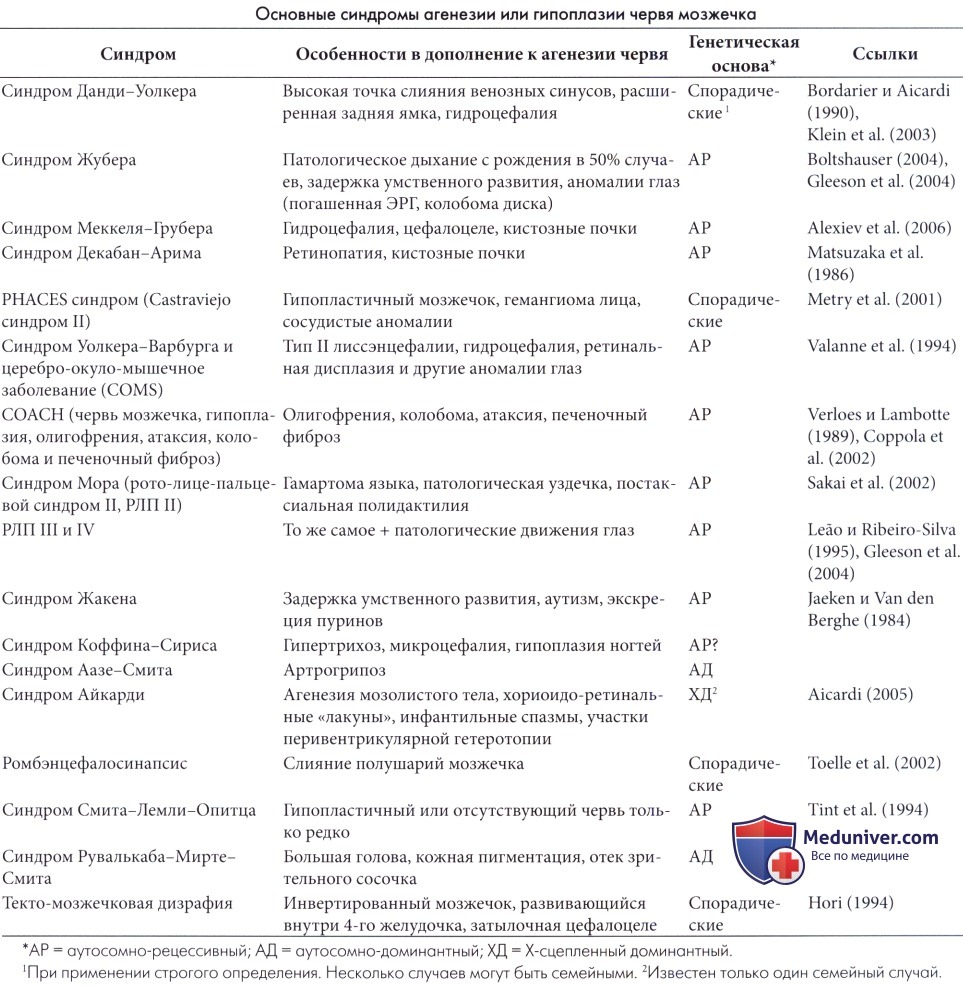
Согласно современным представлениям этиология синдрома Денди-Уокера чрезвычайно гетерогенна, так как в его возникновении принимают участие разные факторы: наследственные (хромосомные и генные) и экзогенные тератогены; предполагается, что определенную роль играют и такие факторы как вирусная инфекция (ЦМВ, краснуха); приём алкоголя, диабет беременно. В 1/3 – 1/2 случаев синдром Денди-Уокера сочетается с другими различными врожденными синдромами (табл. 1) [2].
В 70% случаев порок сочетается и с другими аномалиями головного мозга — агенезией мозолистого тела, энцефалоцеле, полимикрогирией, агирией, гетеротопией серого вещества, а также с поражениями других органов и систем (полидактилией, синдактилией, врожденными пороками сердца, поликистозом почек, расщелинами неба и др.) [5].
Среди основных гипотез возникновения синдрома Денди-Уокера можно выделить следующие:
— остановка эмбрионального развития в процессе формирования ромбовидного мозга;
— атрезия выходного отверстия из IV желудочка при отсроченном открытии отверстия Мажанди;
— возникновение сосудистого сплетения IV желудочка в середине тонкой крыши ромбовидного мозга.
Лечение данного заболевания симптоматическое. При наличии признаков нарастающей внутричерепной гипертензии проводят шунтирующие операции. Исход часто летальный, в 90% случаев это первые годы жизни [7].
Картина пренатального УЗИ в 35 недель гестации: форма головы аномальная (клубникообразная). Кости при надавливании датчиком не деформируются. Расширение боковых желудочков, передние рога до 10 мм справа и слева. Полость прозрачной перегородки до 9,9 мм. Задние рога – 11 мм, слева – 12 мм. Отмечено расширение IVжелудочка до 12 мм. Имеется гипоплазия червя мозжечка, расширение большой цистерны до 12 мм.
Состояние ребёнка после рождения тяжелое, из родильного зала поступил в ОРИТН, за счёт дыхательной недостаточности III степени и выраженной неврологической симптоматики. C момента поступления находился на ИВЛ, с третьих суток жизни был переведен на респираторную поддержку в режиме СРАР, а с 7 суток жизни на спонтанное дыхание. Оксигенотерапия проводилась по показаниям. Энтеральное питание начато с первых суток в трофическом объеме с последующим расширением, усваивал.
Неврологически: положение ребёнка вынужденное на боку с запрокинутой головой, крик монотонный, имеются плавающие движения глазных яблок, симптом «заходящего солнца», непостоянный горизонтальный нистагм. Неправильная форма черепа, увеличенная в размерах мозговая часть, нарастание окружности головы в динамике за 2 недели составило 2,5 см. Отмечалось расхождение костей черепа, швы открыты до 0,5 см, с увеличением размеров большого родничка до 5 × 8 см. Зрачки средней величины, фотореакция сохранена. Спонтанная двигательная активность и мышечный тонус снижены. Дистония в конечностях. Физиологические рефлексы угнетены, сухожильные снижены, симметричные.
Обращало на себя внимание наличие двусторонней косолапости с варусной деформацией обеих стоп, кистей и фаланг пальцев, гипертелоризма, седловидной переносицы, нависающего лба и затылка, широкого языка, короткой шеи (рис. 3).
На 14 сутки жизни, после стабилизации состояния, ребёнок был переведён на второй этап лечения, в отделение патологии новорожденных для дальнейшего наблюдения и лечения.
Картина нейросонографии в первые сутки жизни: в задней черепной ямке при коронарных и сагиттальном сканированиях наблюдается крупное анэхогенное («кистовидное») образование, включающее расширенные III и IV желудочек; полушария мозжечка резко уменьшены, червь не определяется; мозжечковый намет смещен вверх.
В динамике по данным НСГ выявлена перивентрикулярная ишемия, подтверждён синдром Денди-Уокера, вентрикуломегалия (как часть симптомокомплекса) и повышенная резистентность сосудов мозга.
На момент осмотра в нейрохирургическом лечении не нуждался, были даны рекомендации по уходу и лечению.
По результатам ДЭХО-КГ выявлен врожденный порок сердца: комбинированный стеноз легочной артерии (клапанно-подклапанный), ДМПП со сбросом слева направо. Открытое овальное окно диаметром 2,0 мм.
Ребенок был консультирован кардиологом и кардиохирургом, даны рекомендации.
По данным УЗИ брюшной полости и почек патологии не выявлено.
После осмотра врача-ортопеда была подтверждена врожденная двусторонняя косолапость.
В связи со стабилизацией состояния ребёнка, после проведённого обследования и лечения, а также отказа матери от родительских прав, мальчик на 57 сутки жизни был переведён в дом ребёнка города Маркса.
В заключение следует сказать о том, что проведённое настоящее наблюдение представляет большой интерес с клинической точки зрения, поскольку встречается ни так часто в повседневной практике врача.
Ранняя диагностика сложных генетических синдромов, к коим относится и описываемое клиническое наблюдение, представляет определенные сложности. По моему мнению, в подобных ситуациях оправдана постановка синдромального диагноза с уточнением аномалий развития на основании анализа совокупности клинических данных, дополнительных методов обследования. Точный нозологический диагноз важен не только для генетического анализа, медико-генетического консультирования, но и, прежде всего, для профилактики и лечения. Без достоверного клинического диагноза невозможен анализ факторов и их теоретическое осмысление.
Литература
1. Кириллова Е.А., Никифорова О.К., Жученко Н.А. и др. Мониторинг врожденных пороков развития у новорожденных // Российский вестник перинатологии и педиатрии. — 2000. — № 1. — С. 18-21.
2. Барашнев Ю.И. Перинатальная неврология. – М.: Триада-Х, 2001.- С. 181-232.
3. Бочков Н.П. Наследственные болезни. Национальное руководство. М.: ГЭОТАР-Медиа, 2012.- С. 128-145.
5. Наследственные болезни / Под ред. Л.О.Бадаляна. – Т.: Медицина, 2002.- С. 138.
6. Пренатальная диагностика наследственных и врожденных болезней / Под ред. Э.К.Айламазяна, В.С.Баранова. – М.: Триада-Х, 2007. – С. 11-148.
7. Барашнев Ю.И., Бахарев В.А., Новиков П.В. Диагностика и лечение врожденных и наследственных заболеваний у детей. М.: Триада-Х, 2004.- С.12-87.
Таблицы
Синдромы, с которыми сочетается гидроцефалия Денди-Уокера
Синдромы
Клинические признаки
Этиология
Различные хромосомные аномалии
Поражение различных органов и систем
Хромосомные делеции, дупликации, трисомии, триплоидии
Лиссэнцефалия, ретинальная дисплазия, аномалия глаз, энцефалоцеле, миопатии и др.
Мутации генов POMT1 (локализации 9g34.1), POMT2 (14g24.3), FKTN (9g31) и др. Тип наследования аутосомно-рецессивный
3С синдром (краниомозжечково-сердечная дисплазия, Ritscher-Schinzel синдром)
Задержка роста, пороки сердца (септальные дефекты), гипертелоризм и др.
Ген не установлен. Тип наследования аутосомно-рецессивный
Артериальные аномалии, включая коарктацию аорты, дефекты сердца, глаз (микроофтальмия).