Прионовая болезнь что это
Прионовые болезни – это прогрессирующие нейродегенеративные заболевания с летальным исходом. Механизм возникновения прионовых болезней изучен не до конца, однако в настоящее время считается, что заболевание развивается при накоплении в клетках центральной нервной системы избыточного количества патологического прионного белка.
Прионовые болезни встречаются очень редко – ежегодно регистрируется один случай на миллион человек. На данный момент известны 5 разновидностей этой патологии – спорадическая болезнь Крейцфельда-Якоба, новый вариант болезни Крейцфельда-Якоба, куру, синдром Герстманна-Штройслера-Шейнкера и фатальная семейная инсомния. Наиболее широко распространена спорадическая болезнь Крейцфельда-Якоба.
Болезнь может возникать попадании в организм патологического прионного белка извне (от человека или животного), иметь наследственную природу или возникать спонтанно как следствие генетических аномалий de novo. Течение определенных вариантов зависит от типа прионного белка. Инкубационный период может достигать 15 лет. Продолжительность жизни пациентов с прионными болезнями чаще всего составляет около года, реже – чуть более двух лет. Лечениесимптоматическое.
Трансмиссивная губчатая энцефалопатия.
Human prion diseases, transmissible spongiform encephalopathies, TSE.
Симптомыпатологии могут возникнуть в период от полугода до 10-15 лет после заражения. Проявления болезни могут развиваться постепенно, с неспецифических симптомов – бессонницы, вялости, апатии, заторможенности. В ряде случаев болезнь начинается внезапно и может напоминать делирий. Наиболее частыми проявлениями прионовых болезней являются:
Все проявления прионных болезней неуклонно прогрессирую, приводя в конечных стадиях болезни к акинетическому мутизму и коме.
Общая информация о заболевании
Прионные болезни встречаются как у человека, так и среди животных. Патогенез прионных болезней связывают с накоплением в организме патологического прионного белка. Прионный белок в норме присутствует в клетках организма животных и человека – это так называемый нормальный прионный белок. Он находится в наибольших количествах в нейронах и частично в клетках лимфоидной ткани. Прионный белок устойчив к высоким температурам, радиации, действию протеаз и химических веществ – алкоголя, формалина и других. Функции прионных белков неизвестны. Информация о структуре этих полипептидов закодирована в коротком плече 20 хромосомы у человека.
При изменении конфигурации нормальный прионный белок может превращаться в патологический. Патологический прионный белок, в свою очередь, способен запускать механизм преобразования нормального прионного белка в патологическую форму. Патологический прионный белок может возникать в организме спонтанно. В некоторых случаях накопление патологического прионного белка связано с наследственными генетическими нарушениями. На данный момент известно порядка 30 вариантов генетических нарушений, в результате которых меняется структура прионного белка. В ряде случае прионные болезни носят инфекционную природу. Это означает, что патологический прионный белок может попасть в организм человека извне – при пересадке роговицы, твердой мозговой оболочки, нейрохирургических операциях. В данном случае большое значение имеет устойчивость прионов к любым видам стерилизации. Известны случаи заражения патологоанатомов. Считается, что существует вероятность заражения человека при употреблении мяса коров, зараженных коровьим бешенством.
Несмотря на то, что часть прионных заболеваний имеет инфекционную природу, прионы отличаются от других инфекционных агентов – в их составе отсутствуют нуклеиновые кислоты (ДНК или РНК). Это осложняет процесс изучения патогенеза прионовых болезней. Известно, что накопление патологического прионового белка приводит к разрушению клеток нервной системы, мультифокальным спонгиоформным (губкоподобным) повреждениям тканей нервной системы, астроглиозу при отсутствии признаков воспалительной реакции. В связи с устойчивостью к действию протеаз, патологический прионный белок не может быть выведен из организма. Около 85 % пациентов погибают в течение года после появления первых симптомов болезни.
Кто в группе риска?
К сожалению, на данный момент диагностика прионных болезней возможна лишь после возникновения клинических проявлений, то есть на этапе, когда болезнь зашла уже достаточно далеко. Раннее, пресимптоматическое выявление данной патологии невозможно. Схема диагностики прионных болезней не разработана окончательно.
Патогенез прионных заболеваний состоит из двух стадий — инфекционной и токсической

Патологический прионный белок, попадая в нейрон, заставляет неправильно складываться нормальные прионы; переродившиеся белки слипаются в смертоносные агрегаты и убивают клетку. Выяснилось, что количество плохих прионов растет до одного и того же уровня независимо от исходного количества хороших, зато следующая фаза болезни — фаза плато — тем короче (и тем быстрее наступает смерть), чем больше хороших прионов было в клетке.
Прионные заболевания (см. прионы) — один из самых коварных и загадочных типов нейродегенеративных болезней. Сюда относятся болезнь Крейтцфельдта–Якоба, от которой страдают люди; коровье бешенство, которое поражает крупный рогатый скот (и которое напугало человечество 20 лет назад, когда в Великобритании началась эпидемия этой болезни); скрейпи, от которой умирают козы и овцы; страшное наследственное заболевание человека под названием фатальная семейная бессонница и некоторые другие болезни.
Но самое ужасное то, что, оказавшись в клетке, патологический, вредоносный PrP Sc заставляет «хороший» PrP C сложиться «по-своему», неправильным образом. В результате в клетке нарастает количество неправильно сложенных белков, которые слипаются в смертоносные агрегаты и убивают ее.
Нейроны гибнут; неправильно сложенные PrP Sc из мертвых клеток заражают живые и их тоже доводят до смерти. Всё это приводит к тяжелым поражениям определенных областей мозга, и через некоторое время организм погибает.
Патогенез всех прионных заболеваний состоит из длинного инкубационного периода (у человека он может длиться до 50 лет!), во время которого не проявляется никаких признаков болезни; вслед за ним идет короткая (обычно в несколько месяцев длиной) клиническая фаза, когда уже невозможно ничего сделать. Надо отметить, что у разных людей инкубационный период может занимать совершенно разное время. Что конкретно происходит на этих двух стадиях заболевания в нейронах и с чем связано столь резкое проявление болезни, до настоящего времени было изучено слабо.
Однако в 2003 году такая методика была предложена. Исследователи из Института неврологии Университетского колледжа в Лондоне использовали ее, чтобы понять, что происходит в нейроне, зараженном неправильно сложенными прионными белками.
В своих экспериментах они использовали несколько инбредных линий мышей:
1. Prnp null (Prnp o/o ), не экспрессирующие PrP C вовсе.
4. Tg20, экспрессирующие уровень PrP C в восемь раз больше нормального.
Экспериментальные группы мышей каждой линии были заражены прионным заболеванием, а контрольные получили «пустой укол»; после этого исследователи регулярно измеряли уровень инфекционных частиц в нейронах подопытных животных и следили за их состоянием.
Но когда исследователи попробовали измерить уровень прионов в мозге больных мышей, то результаты оказались гораздо более интересными.
Выяснился еще один интересный момент. Оказалось, что уровень PrP C у мышей обратно пропорционален продолжительности фазы плато (рис. 2). Для фазы роста такой зависимости не было выявлено, хотя небольшие различия по ее продолжительности между тремя линиями всё-таки наблюдались.

Результат, мягко говоря, неожиданный. Гораздо логичней было бы, если бы фаза роста коррелировала с количеством PrP C в клетках (чем больше вокруг «хороших» прионов, тем быстрее они превращаются в «плохие»), а фаза плато, наоборот, нет (тогда бы можно было объяснить ее появление тем, что количество «плохих» прионов в клетке достигло максимально возможного, и теперь они постепенно будут разрушать все вокруг).
Но то, что после достижения прионами фазы плато животные живут разное время, ставит всё с ног на голову. Эти результаты объяснить трудно, если только.
Патогенез болезни тогда выглядит так: в клетку попадает патогенный PrP Sc ; окружающие молекулы PrP C меняют конформацию и сами превращаются в PrP Sc (на графике это выглядит как фаза роста); затем достигается максимально возможный уровень PrP Sc (начинается фаза плато), теперь уже PrP Sc запускает образование PrP L из PrP C (фаза плато продолжается). Длительность фазы плато обратно пропорциональна количеству в клетке PrP C — если его много, то она будет короткой, если мало — наоборот, длинной. Иными словами, болезнь состоит из двух стадий — инфекционной (когда «работает» PrP Sc ) и токсической (когда за дело берется PrP L ).
В пользу этого предположения говорит многое — прежде всего то, что, действительно, помимо двух хорошо изученных конформаций прионного белка существует еще множество слабоисследованных. (Вполне возможно, что на самом деле в патогенезе участвуют даже не три, а больше форм белка.) Два главных автора обсуждаемой статьи еще в 2007 году высказывали сходную теорию, и эти результаты служат отличным подтверждением их идей.
Однако возможны и другие интерпретации полученных данных. Может быть, всё дело просто в особенностях измерения концентрации PrP Sc (методика появилась недавно, и многие ее подводные камни еще не известны). Кроме того, показано, что PrP С служит рецептором для PrP Sc и за счет запускаемых в результате их взаимодействия каскадов реакций в клетке возникают токсические эффекты. Если дело в этом, то понятно, почему продолжительность фазы плато зависит от уровня PrP С — чем его больше, тем быстрее «работает» PrP Sc и тем быстрее доводит клетку до смерти (и тем меньше длится фаза плато). В любом случае, полученные результаты очень интересны и явно требуют дальнейших исследований.
Источник: Malin K. Sandberg, Huda Al-Doujaily, Bernadette Sharps, Anthony R. Clarke, John Collinge. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases // Nature. 24 February 2011. V. 470. P. 540–542.
Прионы: идеальные убийцы и возможный ключ к бессмертию
Фото: M24.ru/Евгения Смолянская
За свою историю человечество сталкивалось с огромным количеством войн, эпидемий, стихийных бедствий и других катаклизмов. В XXI веке, когда с эпидемиями, казалось, было покончено, у человечества появился новый вызов – прионы. Что это такое, чем они грозят людям и почему прионами так интересуются ученые всего мира – в материале M24.ru.
Ты помнишь, как все начиналось
В двадцатые годы прошлого столетия врачи столкнулись с новым и неизведанным доселе заболеванием. Немецкий невропатолог Ганс Герхард Крейтцфельдт наблюдал в своей клинике одну пациентку – 20-летнюю девушку. На начальной стадии болезни у нее была нарушена чувствительность в руках и ногах, быстро прогрессировали расстройства памяти, нервной деятельности, больная все чаще впадала в бессознательное состояние. Через несколько месяцев девушка умерла от расстройств дыхания и сердечной деятельности. Невропатолог, который в будущем станет видным нацистским врачом и будет принимать участие в программе «Эвтаназия», задокументировал ход болезни.
Спустя несколько месяцев доктор Альфонс Мария Якоб из Гамбурга столкнулся с тремя аналогичными пациентами. Молодые люди страдали от расстройств нервной деятельности, глотания, практически не осознавали происходящее вокруг и вскоре умерли. При вскрытии Якоб увидел интересное явление, которое раньше врачам наблюдать не приходилось, – поражен у больных был только мозг. Была зафиксирована массовая гибель клеток серого вещества головного мозга, а сохранившиеся нейроны отличались необычным набуханием. Ни в одном другом органе не было зафиксировано никаких патологических изменений. В память о двух первооткрывателях заболевание получило название болезни Крейтцфельдта – Якоба.
В те далекие годы вирусология как наука находилась еще в зачаточной стадии. Поэтому заболеванию было суждено долгое время оставаться в забвении. Этому поспособствовали Великая депрессия и Вторая мировая война. И лишь в пятидесятые годы прошлого века ученые начали активно интересоваться, что же все-таки происходит с людьми, которым не посчастливилось подхватить болезнь Крейтцфельдта – Якоба.
В то же время ученые открывают еще два заболевания, которые по своим симптомам и течению весьма и весьма напоминают описанный выше страшный недуг – куру и скрейпи. Первая болезнь была распространена среди народности форе на острове Папуа – Новая Гвинея, а вторым страдали овцы по всему миру. Но важным оказалось другое: симптомы болезней несколько отличались от болезни Крейтцфельдта – Якоба, но характер поражений был практически идентичен – образование пустот в тканях головного мозга и массовая гибель нервных клеток.
Казалось бы, все ясно. Имеется болезнь, ее вызывает какой-то вирус или бактерия, давайте разберемся, кто является возбудителем и устраним причину. Но не тут-то было! Все оказалось не так просто.

«Познавательные фильмы»: Вакцины
Исследования
Ученым удалось достаточно быстро установить, почему болеют папуасы. Выяснилось, что заболевают только те из них, кто участвовал в ритуальном поедании тел погибших от куру родственников. Согласно местным верованиям того времени, дети должны были обязательно отведать мозга умершего, считалось, что от этого у них прибавится ума. Неизвестно, прибавлялось ли у детей от этого ума, но все малолетние, участвовавшие в таких трапезах, обязательно оказывались зараженными куру.
Особенно масштабные исследования развернулись с агентом скрейпи. Для начала определили его размеры, они оказались стандартными для вирусов – 17–27 нанометров. После этого вирусологи всего мира стали разбираться в свойствах неизвестного возбудителя заболевания, и тут их ждали сюрпризы. Оказалось, что инфекционный агент совершенно невосприимчив к формалину, пепсину и трипсину, не реагирует на ферменты, разрушающие ДНК и РНК, устойчив к кипячению, ультрафиолетовому излучению и. проникающей радиации! С такими вирусами ученым сталкиваться еще не приходилось.

Фото: M24.ru/Александр Авилов
Больше того, возбудителя заболевания никак не удавалось увидеть в электронный микроскоп, что было уж совсем странно. В то время ученые уже умели распознавать вирусные частицы намного мельче, чем 17 нанометров, но вирус скрейпи (почесуха) так никто и не увидел – наблюдали лишь фрагменты клеточных мембран.
Еще одной интересной загадкой оказалось всякое отсутствие иммунного ответа организма больных. Организм людей, больных куру, и овец, страдавших от скрейпи, никак не реагировал на течение заболевания. При обычных болезнях, вроде гриппа и простуды, в организме увеличивается синтез интерферона (отвечает за иммунитет), что ведет к быстрому выпуску антител, которые соединяются с вирусными частицами и растворяют их. Ученые пытались обнаружить признаки хоть каких-либо антител, но потерпели неудачу.
Отчаявшиеся исследователи начали выдвигать гипотезы, что возбудителем является не вирус, а молекула полисахарида или же белка, но подтверждения эта версия так и не нашла. Ученые топтались на месте, пока в 1982 году американский невролог Стэнли Прузинер не заявил об открытии нового класса инфекционных агентов – прионах.
Что такое прион
До открытия прионов считалось, что болезни человека и животных могут вызываться исключительно живыми организмами или хотя бы вирусами, содержащими нуклеиновую кислоту. Однако все оказалось не так просто. Прион – это особый вид белка, который присутствует в любом человеческом организме.
Выяснилось, что либо под воздействием непонятных факторов, либо из-за мутаций в организме некоторых людей нормальный прионный белок, входящий в состав клеточных мембран, заменяется «неправильным». Второй вид прионного белка имеет другую структуру, вызывает гибель клеток, но самое интересное – способен самостоятельно размножаться (без каких-либо ДНК и РНК!) и менять нормальные прионы в соседних клетках на дефектные.

«Познавательный фильм»: Вирусы и защита от эпидемий
Таким образом, прионы оказались единственным видом инфекционных агентов, которых никак нельзя причислить к живым существам. Ведь, по своей сути, они не содержат никакой генетической информации и самостоятельно синтезируются организмом.
Естественно, исследователей заинтересовал самый главный вопрос – а зачем вообще в человеческом организме нужны прионы? В настоящее время известно уже достаточно много прионных болезней. Все они являются экстремально редкими, самая распространенная – болезнь Крейтцфельдта – Якоба – наблюдается у одного из миллиона человек. Также известно о синдроме Герстманна – Штраусслера – Шайнкера, фатальной семейной бессонице и куру. Некоторые исследователи включают в группу прионных заболеваний человека также болезнь Альперса у детей, амиотрофический лейкоспонгиоз (описан белорусскими учеными в конце прошлого века, болели работники одной из ферм) и спонгиоформный миозит (мышечное истощение).
Все эти заболевания являются смертельными, и лекарств от них пока не предложено. Но все же зачем организм синтезирует прионы? Какую он отводит роль для них?
Зачем нужны прионы?
В 70-е годы прошлого века два английских исследователя – Паттисон и Джебет – изучали на мышах действие вещества под названием купризон. В нормальных условиях оно связывает в организме ионы меди. Животным включили купризон в обязательную диету с целью посмотреть, какое действие он произведет на грызунов. И поразились! После 30 с лишним дней купризоновой диеты совершенно здоровые до этого мыши превратились в тяжелобольных. Причем все признаки заболевания полностью отвечали симптомами скрепи. Часть мышей, участвовавших в эксперименте, вскрыли и посмотрели – оказалось, что в головном мозгу животных произошли абсолютно те же изменения, что и при прионных болезнях.
Возник вопрос: а что если купризон мышам больше не давать? Попробовали – и через несколько дней грызуны выздоровели. Уже через 30 дней у них исчезли и вызванные купризоном изменения в мозговой ткани.
Спустя много лет было выяснено, что прионы весьма и весьма похожи на положительно заряженные частицы двухвалентной меди. И изменения, которые они вызывают в организме, практически идентичны. Таким образом, исследователи сделали вывод о том, что в нормальном состоянии прионы отвечают за оборот металлов, в частности меди. Но эти данные пока остаются лишь гипотезой.

Фото: ТАСС/Станислав Красильников
Еще одна группа американских исследователей принялась копать в другом направлении. Им удалось получить данные, что прионы помогают клеткам мозга прикрепляться друг к другу и участвуют в передаче сигналов внутри клетки. Это означает, что отсутствие прионов или их дефекты не позволяют клеткам мозга получать сигнал о других клеток, что ведет к развитию тяжелых нарушений в работе нервной и других систем организма.
Но самым интересным является предположение о том, что прионы участвуют в механизмах клеточного старения. Не секрет, что долгое время прионные болезни относили к группе старческих болезней, потому что вызываемые ими изменения весьма сходны с другими заболеваниями (вроде болезни Пика, Альцгеймера и других неврологических недугов). Наличие прионной инфекции как бы подталкивает организм к ускоренному старению. Естественно, это ставит очень важный вопрос: если лекарство от таких болезней будет найдено, не станет ли оно своеобразным ключом к долголетию или даже бессмертию организма? Но ответ на этот вопрос пока дать невозможно, поскольку функции прионов изучены еще недостаточно хорошо.
Способы заражения
В заключение поговорим о способах заражения. Их четыре. В первом и самом распространенном случае заболевание возникает как бы из ниоткуда. То есть жил себе человек, да вдруг взял и заболел. Этот путь возникновения болезни называется спорадическим и, кстати сказать, является наиболее распространенным. По нынешним представлениям, это происходит спонтанно под действием каких-то пока не установленных факторов.
Второй способ – наследственный. Некоторые виды болезней являются семейными и возникают из-за мутаций. В свою очередь, гены передаются потомству. Известно около 40 семей, страдающих фатальной бессоницей. Каждый десятый страдающий болезнью Крейтцфельдта – Якоба – страдает семейной формой этого заболевания.
Фото: M24.ru/Михаил Сипко
Третий способ – ятрогенный. Это означает, что заражение прионами произошло по вине медицинских работников при проведении каких-либо оперативных вмешательств. Однако описаны лишь несколько таких случаев, и все они произошло в 70-е годы прошлого века, когда о свойствах прионов еще никто не знал. Так, одна женщина заболела после того, как ей пересадили роговицу глаза от страдавшего болезнью Крейтцфельдта – Якоба мужчины.
А вот последний способ наиболее коварен и опасен. Дело в том, что человек восприимчив к прионам, которые поражают крупный рогатый скот. И при употреблении в пищу мяса больных животных заболевают и люди – у них развивается болезнь Крейтцфельдта – Якоба. В девяностые годы прошлого века настоящая эпидемия этого страдания разразилась в Англии.
Лечения пока нет. Однако ученые уже выяснили, что некоторые виды прионов разлагаются лишайниками, другим удалось описать особые антиприонные антитела (к инфекционным прионам).
Иными словами, перед исследователями стоит весьма непростая задача, которая не только поможет найти лекарство от тяжелых заболеваний, но и, возможно, поможет открыть секрет долголетия. Для этого нужно только одно – понять прионы.
Прионы: исследования таинственных молекул продолжаются
Путь прионов
Автор
Редакторы
Статья на конкурс «био/мол/текст»: Прионные заболевания — феномен, открытый в двадцатом веке, и в нем же начавший играть большую роль: увеличение продолжительности жизни в развитых странах привело к тому, что все больше людей стало доживать до «своего Альцгеймера» или «своего Паркинсона». Природа нейродегенеративных заболеваний продолжает оставаться туманной, и ученые пока исследуют только отдельные их аспекты — например, причину развития именно в старческом возрасте или способность передаваться от одних видов живых существ другим.

«Био/мол/текст»-2012
Эта статья представлена на конкурс научно-популярных работ «био/мол/текст»-2012 в номинации «Лучшее новостное сообщение».
Спонсор конкурса — дальновидная компания Thermo Fisher Scientific.
Всё началось с того, что в 20 веке учёные заинтересовались природой необычных заболеваний человека и животных: куру, Крейтцфельда-Якоба, скрэпи. Заметное сходство патологии этих болезней дало основание для гипотезы об их инфекционности, что впоследствии было экспериментально подтверждено. Тогда возник вопрос о возбудителе данных заболеваний. Прежде чем был найден ответ, были выявлены необычайные свойства возбудителей: они не размножаются на искусственных питательных средах, устойчивы к высокой температуре, формальдегиду, различным видам излучений, действию нуклеаз. Очистка инфекционного материала и его изучение позволило провозгласить о том, что «во всём виноват» белок, который 30 лет назад получил название прион (от англ. pr[otenacious infect]ion — белковая инфекция).
Так, известные американские учёные — вирусолог и врач Д.К. Гайдушек, раскрывший инфекционную природу прионных болезней, в 1976 г. и биохимик С.Б. Прузинер, который определил прионы и разработал прионную теорию, в 1997 г., — были удостоены Нобелевских премий. Их работы стали импульсом для последующих исследований, благодаря которым были изучены новые виды прионных инфекций. Но, даже несмотря на неугасающий интерес к «прионной теме», образование прионов до сегодняшнего дня остаётся загадкой.
Биологическая сущность прионов
Рисунок 1. Метафора нейродегенеративного поражения мозга — это губка, в которую превращается нервная ткань в результате массовой гибели нейронов.
Существует упрощенное представление, что полимеризованные прионные фибриллы «протыкают» нейрон, что вызывает его гибель. На самом деле это не совсем так: предшествующие фибриллярной стадии сферические агрегаты прионов также обладают токсичностью (по крайней мере, для болезни Альцгеймера): «Альцгеймеровский нейротоксин: ядовиты не только фибриллы». — Ред.
Но как может нормальный природный белок (обозначается PrP C ) вдруг стать патологическим (PrP Sc ; Sc — от слова «scrapie»)? Что должно произойти? Как и в случае «обычной» инфекции, для такой трансформации необходима встреча с молекулой инфекционного приона. Существуют два пути передачи этой молекулы: наследственный (за счёт мутаций в гене, кодирующего белок) и инфекционный. То есть внедрение приона может произойти неожиданно — например, при употреблении в пищу недостаточно хорошо прожаренного или сваренного мяса (в котором должна присутствовать форма PrP Sc ), при переливании крови, при трансплантации органов и тканей, при введении гормонов гипофиза животного происхождения.
И тогда происходит удивительное событие: нормальные молекулы белка, контактируя с патологическими, сами превращаются в них, изменяя свою пространственную структуру (механизм трансформации остаётся загадкой и по сей день) [1]. Таким образом прион, как самый настоящий инфекционный агент, заражает нормальные молекулы, запуская цепную реакцию, разрушительную для клетки.
Некоторые сведения о прионах
Условия возникновения заболеваний
Условия возникновения прионовых болезней уникальны. Они могут формироваться по трём сценариям: как инфекционные, спорадические и наследственные поражения. В последнем варианте главную роль играет генетическая предрасположенность [2].
Знаменитый исследователь прионов Стэнли Прузинер (Stanley Prusiner) выделяет две поразительные особенности, присущие таким нейродегенеративным заболеваниям, как болезнь Крейтцфельда-Якоба, болезнь Альцгеймера и болезнь Паркинсона. Первая заключается в том, что более 80% случаев заболевания — спорадические (то есть, случайные, возникающие «сами собой»). Вторая: несмотря на то, что большое количество мутантных белков, специфичных к определённой болезни, экспрессируется в процессе зародышевого развития, формы наследования этих нейродегенеративных заболеваний проявляются позже. Это предполагает, что некоторые процессы происходят во время старения, которое «дает волю» болезнетворным белкам [5]. Более 20 лет назад автор утверждал, что данный процесс включает случайный рефолдинг (пересворачивание) белка в неправильно свёрнутый, что соответствует переходу в инфекционное состояние — прион.
Интересные факты насчет болезни Альцгеймера: ее вероятность может повышаться вследствие хронического недосыпания («Новый шаг к пониманию болезни Альцгеймера: возможно, недосыпание является одним из факторов риска»), а сам альцгеймеровский нейропептид (β-амилоид Aβ) может быть частью системы врожденного иммунитета («Возможно, β-амилоид болезни Альцгеймера — часть врождённого иммунитета»). — Ред.
В последнее десятилетие интерес к этой теме возобновился в связи с возможностью развития диагностики и эффективной терапии [5]. Появилось множество различных объяснений для возрастных нейродегенеративных болезней, — например, окислительная модификация ДНК, липидов и/или белков; соматические мутации; измененный врождённый иммунитет; экзогенные токсины; несоответствия ДНК—РНК; нарушение работы шаперонов; отсутствие одного из аллелей гена [5]. Альтернативным комплексным разъяснением служит то, что различные группы белков могут формировать прионы. Несмотря на то, что небольшое количество прионов может быть удалено посредством путей белковой деградации, их чрезмерное накопление с течением времени позволяет прионам самостоятельно распространяться в организме (рис. 2), что приводит к нарушению деятельности центральной нервной системы [5].

Рисунок 2. Процессы нейродегенерации, вызванной прионами. Сверху: накопление «нормального» прионного белка повышает его вероятность перехода в токсичную конформацию, которая описывается бóльшим содержанием β-структуры. Прионы наиболее патогенны в форме олигомеров; после образования фибрилл токсичность снижается. В зависимости от того, о каком конкретно прионном белке идет речь, в патологическом состоянии он может образовывать бляшки, клубки или тельца включения. Возможные пути лекарственного вмешательства: (I) снижение концентрации «нормального» белка-предшественника; (II) ингибирование образования прионной формы; (III) уничтожение токсичных агрегатов. Снизу: Наследственная старческая нейродегенерация объясняется двумя событиями: наличием мутантной формы предшественника и образованием из него приона, готового к олиго- и полимеризации с образованием токсичных форм.
Группы риска прионных заболеваний
Вот кого прионные заболевания могут настичь с наибольшей вероятностью:
Лабораторная диагностика и лечение
Диагностика базируется на внутримозговом заражении мышат или хомяков, у которых медленно (до 150 дней) развивается соответствующее заболевание, если пациент был болен [2]. Часто проводится гистологическое исследование головного мозга погибших животных [2].
К сожалению, до настоящего времени еще не разработаны эффективные методы лечения прионовых болезней, хотя попытки предотвратить конформационный переход нормального белка в аномальный производятся. Поэтому самым надёжным способом предупреждения развития инфекционных форм является профилактика [2].
Особенно актуальным становится решение «прионного вопроса» в связи с нарастающей угрозой возникновения эпидемии через инвазивные медицинские операции и даже при приёме лекарственных средств.
Перспективы
По-видимому, интерес к прионам не угаснет до тех пор, пока предположения на их счёт полностью не подтвердятся и не будут найдены эффективные способы лечения прионных заболеваний. В статье [6] говорится о необходимости современного исследования, которое требует тщательного рассмотрения чужеродных прионов в экстраневрональных тканях.
В качестве модельных объектов авторы использовали мышей: две линии, которые трансгенно экспрессировали овечий прионный белок, и одну линию, которая экспрессировала человеческий прионный белок (рис. 3). Задачей было сравнить эффективность межвидовой передачи инфекции посредством тканей мозга и селезёнки. Внутримозговое заражение чужеродным прионным белком выражалось в отсутствии или небольшом количестве инфекционного агента в мозгах этих мышей. Однако инфекционные чужеродные прионы обнаруживались в селезёнке на более ранних этапах заражения в сравнении с моментом, когда были использованы нейротропные прионы, тем самым определяя, что лимфатическая ткань может быть более пермиссивной к распространению чужеродных прионов по сравнению с мозгом.
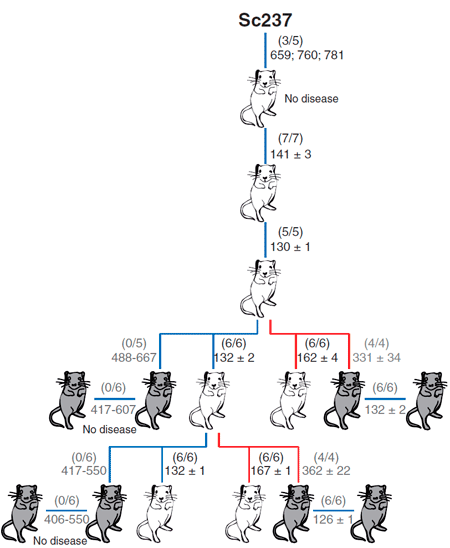
Рисунок 3. Способность приона хомяков Sc237 заражать и передаваться при введении в мозг или селезенку трансгенным мышам, имеющим прионный белок PrP овцы (tg338; белые мыши) или человека (tg7; серые мыши). Число заболевших/инъецированных мышей показано в скобках; ниже приведено среднее время жизни (в днях).
Чем вызвана эта предпочтительная репликация прионов в лимфатических тканях, пока неизвестно. Однако полученные данные показывают, что человек может быть более чувствительным к чужеродным прионам, чем предполагалось ранее на основании присутствия прионов в мозгу, и по этой причине бессимптомный переносчик прионной болезни может быть не распознан. Это ещё раз подтверждает, что такая могущественная биомолекула как прион таит в себе немало загадок, раскрытие которых, возможно, поможет в понимании ряда неразрешимых проблем человечества.