Полисерозит это что такое
Клиническая картина туберкулезного полисерозита осложняется в связи с образованием массивных и распространенных сращений в различных полостях организма. Кроме того, у части больных он начинается как сухой или серозно-фибрииозный плеврит, перикардит или перитонит. На этой почве возникают перивисцериты — перигастрит, перидуоденит, периспленит, перигепатит, периколит, пери- и параметрит. В подобных случаях отсутствуют симптомы острого перитонита, а заболевание протекает под видом аппендицита, холецистита, цирроза печени и т. д.
Все эти клинические особенности следует учитывать при дифференциальной диагностике туберкулезного полисерозита с подобными заболеваниями другой этиологии. Чаще всего приходится исключать ревматическую природу полисерозита. При этом следует иметь в виду, что при ревматизме полиартрит и плеврит большей частью сочетаются с перикардитом и эпдомиокардитом (панкардит, плевроперикардит). Между тем при туберкулезном полисерозите поражается главным образом плевра и брюшина.
Различен характер начальных проявлений и течения этих заболеваний. Туберкулезный полисерозит может начинаться и протекать остро, подостро или хронически; ревматический полисерозит возникает, как правило, более остро, с ремиттирующей лихорадкой, общей слабостью, потливостью. Экссудат, образующийся в плевральной, перикардиальной и брюшной полостях, рассасывается значительно быстрее при ревматизме, чем при туберкулезе, и при этом не образуются массивные сращения.
В плевральной жидкости при туберкулезе в большем количестве содержатся белок, фибрин, лимфоциты, моноциты, нередко находят микобактерии туберкулеза. В ревматическом экссудате мало фибрина, преобладают нейтрофилы, много эндотелиальных клеток, отсутствуют микобактерии туберкулеза. При туберкулезе, как правило, определяются положительные, часто резко выраженные туберкулиновые реакции. У больных ревматизмом они нередко отрицательные или сомнительные.
При туберкулезном полисерозите в отличие от ревматического, как правило, поражены внутригрудные лимфатические узлы; в первом случае, несмотря на длительное и волнообразное течение болезни, не образуется клапанного порока сердца. При ревматическом эндомиокардите он наблюдается очень часто. При туберкулезном поражении сердца отсутствует эффект от салицилатов, амидопирина или реопирина; он часто наступает при применении туберкулостатических средств, которые не влияют на течение ревматического полисерозита.
Сравнительно редко встречающийся ревматический перитонит отличается также нестойкостью и летучестью абдоминального синдрома. Он возникает обычно вскоре после перенесенной ангины, скарлатины и других заболеваний, сочетается с поражением эндокарда, миокарда и полиартритом. Под влиянием противоревматического лечения признаки перитонита быстро исчезают.
Возможны затруднения при распознавании этиологии перикардита, которая может быть различной. На большом клиническом и секционном материале (3269 случаев поражения околосердечной сумки) А. А. Герке (1950) установил, что у 18% больных перикардит развился па почве нефрита и уремии, у 17% — ревматизма, у 13% — туберкулеза, у 9% — стрепто-стафилококковой инфекции, у 44% — рака, у 3% — инфаркта миокарда и т. д. Sodeman и Smith (1958) среди 240 больных перикардитом у 23% установили его неспецифическую воспалительную природу, у 16% больных процесс был связан с гнойной инфекцией, у 11% — с ревматизмом, у такого же количества больных — с инфарктом миокарда. У 8% больных перикардит был вызван раком и лишь у 7% — туберкулезом.
Установить причину поражения перикарда можно, следовательно, только в результате тщательного и всестороннего исследования больного.
Соответствующая клиническая картина болезни позволяет отличить туберкулезный полисерозит от подобного процесса на почве системной красной волчанки. Как известно, волчаночный полисерозит является составной частью «малой триады», характерной для этого заболевания (артрит, дерматит и полисерозит). В таких случаях также паблюдаются выпотной или слипчивый плеврит, перикардит, перитонит, припухание суставов. Одновременно увеличиваются периферические, а иногда и внутригрудные лимфатические узлы.
Нередко при этом возникает хронический иптерстициальный пневмонит. Заболевают волчанкой преимущественно женщины молодого возраста. Все эти клинические симптомы могут напоминать картину туберкулезного полисерозита. Однако для волчаночного полисерозита характерны, как указывают Е. М. Тареев и соавт. (1965), эфемерность клинических симптомов, быстрое развитие и рассасывание выпота, частые рецидивы процесса, неправильный тип лихорадки, возникновение волчаночного кардита и васкулитов, поражение почек, появление дисковидных ателектазов в базальных отделах легких и высокое стояние купола диафрагмы (синдром Мире). При этом заболевании в крови обнаруживают волчаночные клетки, резко увеличено содержание гамма-глобулинов, оказываются отрицательными туберкулиновые пробы, отсутствует эффект от специфической химиотерапии и оказывают терапевтическое действие кортикостероидные гормоны.
Туберкулезный полисерозит может напоминать состояние, возникающее при сердечной недостаточности, когда в плевральных полостях накапливается транссудат и появляется асцит.
Поражение серозных оболочек наблюдается при метастатическом раке, лимфогранулематозе, бруцеллезе, малярии и т. д. В этих случаях природа болезни может быть распознана на основании других основных ее симптомов.
Ревматоид Понсе необходимо дифференцировать от костно-суставного туберкулеза. Помимо других клинико-рентгенологических симптомов, следует учитывать характерную для ревматоида Понсе множественность поражения суставов, между тем как костный процесс протекает обычно как моноартрит.
Следует иметь в виду, что полисерозит туберкулезной этиологии у взрослых развивается не только в связи с первичной инфекцией. Иногда он образуется и в результате лимфо-гематогенной диссеминации и при вторичном туберкулезе (И. И. Мошковский, 1946), но тогда, как мы наблюдали у ряда больных, отсутствуют признаки поражения лимфатических узлов и гиперсенсибилизации, воспалительный выпот в серозных полостях сравнительно быстро ликвидируется и обычно не рецидивирует.
— Вернуться в оглавление раздела «Пульмонология.»
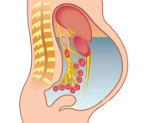
Синдром Мейгса — это особый вариант полисерозита, который возникает у пациенток с опухолями овариальной ткани, матки и полностью проходит после удаления неоплазии. Проявляется увеличением объема живота, нарастанием одышки, тахикардией, слабостью, утомляемостью, бледностью, прибавкой веса при внешних признаках кахексии. Диагностируется при помощи гинекологического осмотра, УЗИ брюшной и плевральной полостей, перикарда, тазовых органов, цитологического исследования плеврального выпота и асцитической жидкости, лапароскопии. Лечение предполагает эвакуацию экссудата, коррекцию органных расстройств, хирургическую экстирпацию опухоли.
МКБ-10
Общие сведения
Синдром Мейгса (Демона-Мейгса, Мейгса-Салмона, Мейгса-Касса) — редкое паранеопластическое расстройство, наблюдаемое у 3% пациенток, страдающих объемными образованиями репродуктивных органов. Симптомокомплекс, проявляющийся асцитом и выпотом в полость плевры у женщин с солидными новообразованиями яичников, был детально описан Дж. Мейгсом в 1934-1935 годах. Несколько позже Р.У. Лайт расширил трактовку синдрома на все новообразования тазовых органов, в том числе злокачественные неоплазии без метастазов. Классическая комбинация овариальной опухоли, асцита и гидроторакса наблюдается в единичных случаях, чаще у пациенток определяется абдоминальный выпот. При наличии плевральной экссудации в 70% случаев процесс локализован справа, в 10% — слева, в 20% — является двусторонним. Расстройство чаще возникает у женщин после 45 лет.
Причины
Патологический симптомокомплекс развивается на фоне неопластического поражения яичниковой ткани и миометрия. Чаще всего полисерозит сочетается с фибромой яичников, овариальными кистами, лейомиомой матки. Плевральный, перитонеальный и перикардиальный выпот также может образовываться при карциноме яичников без признаков метастазирования, хотя в таком случае специалисты в сфере акушерства и гинекологии говорят о псевдосиндроме Мейгса. Описаны спорадические случаи возникновения полисерозита с соответствующей клинической картиной на фоне дегенеративного изменения овариальной ткани без опухолевой трансформации, обширного отека яичников, синдрома их гиперстимуляции гонадотропинами, гонадолиберинами при экстракорпоральном оплодотворении.
Патогенез
Патогенез синдрома Мейгса пока не изучен. Какие-либо специфические каналы, связывающие матку и яичники с полостями плевры и перикарда, не выявлены. Предложено несколько гипотез формирования экссудата при новообразованиях женских репродуктивных органов. Согласно одной из теорий, экссудативный выпот при синдроме Демона-Мейгса-Касса изначально скапливается в брюшной полости в результате возникновения «реакции тревоги» сосудов брюшины на растущую опухоль. Асцит способствует растяжению диафрагмы с формированием конгенитальных дефектов (плевроперитонеальных каналов), по которым экссудат просачивается в плевральную полость.
Ряд авторов не исключает патогенетическую роль лимфатических сосудов, перфорирующих диафрагмальную перегородку. Идея о нарушении венозного и лимфатического оттока в результате механического сдавливания тканей неоплазией не нашла подтверждения, поскольку у части пациенток характерный для синдрома массивный полисерозит развивается при опухолях, диаметр которых не превышает 5 см. Механизмы экссудации также могут запускаться пока неустановленными веществами, секретируемыми новообразованием.
Симптомы
Клиническая симптоматика расстройства нарастает постепенно, является неспецифической и, как правило, становится следствием давления скопившегося выпота на окружающие органы. Пациентка жалуется на периодически возникающую или постоянную незначительную, чаще одностороннюю боль внизу живота, обычно описываемую как дискомфорт. Часть женщин воспринимает болезненные ощущения как тупые, ноющие, распирающие.
В последующем живот увеличивается в размерах, нарастает чувство нехватки воздуха, общее недомогание, слабость, быстрая утомляемость, потливость, ухудшение аппетита, появляется бледность кожных покровов, отечность. Отмечается значительная прибавка в весе на фоне признаков кахектического синдрома (дряблости кожи, мышечной гипотрофии). Уменьшается количество мочи, возможны запоры. У пациенток репродуктивного возраста могут возникать дисфункциональные маточные кровотечения.
Осложнения
При прогрессировании процесса и накоплении значительных объемов экссудативного выпота синдром Мейгса осложняется сердечной и легочной недостаточностью, метаболической кардиомиопатией, анемией, нарастающей ишемией различных органов и тканей. В тяжелых случаях кислородное голодание мозга, вызванное недостаточной оксигенацией крови в легких, способствует возникновению когнитивных расстройств (ухудшения памяти, невнимательности), эмоциональной лабильности, раздражительности, снижению критичности к своему состоянию. На фоне необратимых кахектических изменений возникает полиорганная недостаточность, приводящая к летальному исходу.
Диагностика
Первоначально выпот в брюшной, плевральной, перикардиальной полостях выявляется в ходе физикального обследования (перкуссии и аускультации). О наличии жидкости свидетельствует притупление перкуторного звука над пораженной половиной грудной клетки, в правом и левом фланках живота, расширение границ сердца в обе стороны. Аускультативно везикулярное дыхание в зоне тупости отсутствует или резко ослаблено, сердечные тоны приглушены, учащены. Наличие экссудата подтверждается с помощью рентгенографии грудной клетки, УЗИ плевральной полости, перикарда, органов брюшной полости, эхокардиографии. Выявление перитонеального, плеврального, перикардиального выпота служит основанием для углубленного онкообследования с целью исключения опухолей матки или яичников. Наиболее информативными методами при подозрении на синдром Мейгса являются:
Для выявления опухолевого процесса может быть рекомендована диагностическая лапароскопия, определение онкомаркера СА-125. В общем анализе крови пациенток с синдромом Мейгса обычно несколько снижены количество эритроцитов и уровень гемоглобина, СОЭ увеличено до 25-30 мм/ч, отмечается незначительный лейкоцитоз с палочкоядерным сдвигом влево. На ЭКГ определяются признаки дистрофических изменений миокарда.
Дифференциальная диагностика проводится с экссудативным плевритом, циррозом печени, декомпенсированной сердечной недостаточностью, синдромом полисерозита при туберкулезе, ревматизме, системной красной волчанке, уремии, сепсисе, парапротеинемическом гемобластозе, семейной средиземноморской лихорадке (периодической болезни). По показаниям пациентку консультируют пульмонолог, кардиолог, фтизиатр, ревматолог, нефролог, гематолог, онколог.
Лечение синдрома Мейгса
Терапевтическая тактика направлена на быстрое устранение симптомов компрессии органов, коррекцию сопутствующих расстройств и последующее хирургическое удаление неоплазии. При декомпенсированной полиорганной недостаточности пациентку госпитализируют в отделение интенсивной терапии, в остальных случаях рекомендована плановая предоперационная подготовка в гинекологическом стационаре. Основными этапами лечения синдрома Мейгса являются:
Прогноз и профилактика
Полная резорбция экссудата с восстановлением общего самочувствия обычно происходит в течение 2-х недель после оперативной экстирпации опухоли. У некоторых пациенток остается небольшое количество спаек, плевральных и перикардиальных сращений. При псевдосиндроме Мейгса, осложняющем течение злокачественных новообразований, прогноз зависит от стадии и типа онкологического процесса. Профилактика предполагает плановые осмотры акушера-гинеколога и регулярный УЗИ-скрининг для своевременного выявления и адекватного лечения опухолевого поражения матки, овариальной ткани.
ПОЛИСЕРОЗИТ
П. не самостоятельное заболевание, а синдром различных болезней (инфекционных и неинфекционных), в течении к-рых отмечаются системные реакции. Исключение составляет казуистически редко встречающаяся болезнь Конкато — гиалиноз серозных оболочек неизвестной этиологии.
Частота возникновения и особенности течения П. связаны с основным заболеванием. Напр., одновременное поражение плевральных оболочек и перикарда встречается у половины больных системной красной волчанкой, т. о., синдром П. может иметь определенное диагностическое значение.
Содержание
Этиология и Патогенез
Этиология. Заболевания, при к-рых наблюдается П., многочисленны. Чаще всего П. развивается при системной красной волчанке, ревматизме, ревматоидном артрите, туберкулезе, септических заболеваниях, периодической болезни, а также при уремии, синдроме Дресслера, посткомиссуротомном синдроме. В связи с успехами антибактериальной терапии чрезвычайно редкихМ стал первично-инфекционный П.
Патогенез связан с развитием аллергической, инфекционноаллергической или иммунопатологической реакции. В редких случаях инфекционного П. его патогенез совпадает с общими закономерностями развития инфекционного воспаления (см.) и частными его особенностями, связанными с этиологией (напр., при развитии бугоркового туберкулезного П.). Общность реакций серозных оболочек различной локализации обусловлена единым мезодермальным их происхождением, тесными анатомическими взаимоотношениями, однотипным гистол, строением и схожестью функций. Воспаление серозных оболочек сопровождается их гиперемией, повышением проницаемости обильной субмезоте-лиальной капиллярной сети и выходом плазменного диализата в серозные полости, отрицательное давление в к-рых облегчает этот процесс. В последующем образуется экссудат, содержащий различные клеточные элементы, белок (30 г/л и более), фибрин, протеолитические ферменты и другие компоненты сыворотки крови. Состав экссудата зависит от этиологии и патогенеза основного заболевания, поэтому с диагностической целью используют определение содержания в нем комплемента и его компонентов, глюкозы, иммуноглобулинов различных классов, в т. ч. волчаночного фактора и антииммуноглобулинов (таких, как ревматоидные факторы и агглютинаторы), а также производят микробиол, исследования.
Патологическая анатомия
В морфол, отношении П. разделяют на острый и хронический. Острый П. чаще бывает экссудативным и в зависимости от характера экссудата может быть серозным, фибринозным, реже геморрагическим. Хронический П. развивается, как правило, в исходе острого П. и проявляется хрон, продуктивным воспалением с образованием спаек, реже облитерацией серозных полостей. Особенности морфол, проявлений П. зависят от характера основной болезни.
При ревматизме и сходных заболеваниях (ревматоидном артрите, системной красной волчанке) в серозных полостях обнаруживают серозный, серозно-фибринозный или фибринозный экссудат. Нередко плеврит и перикардит сочетаются с перитонитом (см.); при этом в серозной оболочке, особенно в эпикарде, отмечают выраженную гистио-цитарную реакцию, иногда определяют ревматические гранулемы. Организация экссудата с развитием слипчивого П. приводит к сращению серозных листков и облитерации серозных полостей.
Сходная патологоанатомическая картина наблюдается в ряде случаев П., развивающегося как параспеци-фический процесс при туберкулезе. Собственно туберкулезный П. может возникать гематогенно и в результате распространения процесса по лимф, сосудам в период формирования первичного комплекса. В таких случаях в полостях обнаруживают серозный или серозно-фибринозный экссудат; на плевре, перикарде и брюшине — туберкулезные бугорки, которые могут подвергаться некрозу с последующей организацией и отложением в некротические массы солей кальция. Туберкулезные бугорки на брюшине иногда имеют крупные размеры, что макроскопически может симулировать картину карци-номатоза брюшины. Своеобразный П. с характерной морфол, картиной возникает при уремии.
В исходе острого П. жидкая часть экссудата всасывается, на поверхности серозных оболочек разрастается молодая грануляционная ткань, врастающая в массы выпавшего фибрина. В результате этого происходит утолщение серозных листков, они принимают белесоватый вид, образуются рыхлые или более плотные спайки. В других случаях процесс может иметь хронический продуктивный характер. Резкое очаговое или диффузное утолщение серозной оболочки с превращением ее в плотный белесоватый полупрозрачный панцирь различной толщины называют, по предложению Куршманна (H. Curschmann, 1884), глазурным. Вместе с утолщением серозных оболочек происходит нек-рая деформация покрываемых ими органов. При микроскопическом исследовании на месте серозной оболочки отмечают разрастание гиалинизированной фиброзной ткани со скудными лимфогистиоцитарными инфильтратами между пучками коллагеновых волокон. Тяжи фиброзной ткани обнаруживают в поверхностных отделах паренхимы пораженных органов (печени, селезенки и др., как, напр., при перикардитическом псевдоциррозе Пика).
Клиническая картина
Воспаление нескольких серозных оболочек редко возникает одномоментно; обычно вначале поражается плевра, затем присоединяется перикардит или же наоборот (напр., при остром ревматизме) и соответственно появляются характерные симптомы (см. Перикардит, Плеврит).
Наиболее часто больные жалуются на боли в грудной клетке разной интенсивности и локализации (в боку, за грудиной), боли в верхней половине живота или по всему животу, усиливающиеся при дыхании. Особенно выраженные боли бывают при периодической болезни, остром ревматизме у детей, диафрагматитах при системной красной волчанке. При ревматоидном артрите П. может протекать бессимптомно и нередко обнаруживается только при рентгенол. исследовании или устанавливается при аутопсии.
При развитии П. обычно повышается температура тела. В первые часы и сутки выслушивается шум трения плевры, перикарда, брюшины (над поверхностями печени или селезенки). По мере накопления экссудата шум трения и боли при дыхании умен ып а ют ся или исчезают; при значительных выпотах появляется одышка. Быстрое и значительное накопление экссудата приводит к выраженной одышке, нарушениям кровообращения и появлению симптомов сдавления или смещения экссудатом близко расположенных к нему морфол, образований (органов, сосудов и др.).
В случае инфекционного П. состояние больного может быть крайне тяжелым из-за выраженной интоксикации, высокой лихорадки, скопления в серозных полостях значительного количества экссудата.
П. обычно присоединяется уже к имеющимся клин, признакам основной болезни. При этом обнаруживают характерные для сухого или выпотного П. изменения в отдельных серозных полостях. Однако у детей и подростков при остром ревматизме, ювенильном ревматоидном артрите и нек-рых других заболеваниях П. может быть первым клин, проявлением болезни.
Возникновение П. обычно свидетельствует об активном развитии основного заболевания и сопровождается лейкоцитозом (лейкопенией при системной красной волчанке), нейтрофилезом, значительным ускорением РОЭ (более 40—50 мм/час),.
П. продолжается от нескольких недель до месяца и более и нередко заканчивается формированием спаек (плевродиафрагмальных, плевроперикардиальных), приводящих к неполному раскрытию реберно-диафрагмальных синусов, снижению экскурсии диафрагмы, высокому ее стоянию; на ограниченных участках постоянно выслушивается шум трения пораженной серозной оболочки. При периодической болезни П. склонен к рецидивированию, однако стойкого спаечного процесса не развивается. Слипчивый (сдавливающий) П. чаще всего возникает при туберкулезе и ревматизме и характеризуется медленно прогрессирующей облитерацией серозных полостей фиброзирующими перивисцеритами с нарушением функционирования органов. Рецидивирующий П. при системной красной волчанке приводит к значительному нарушению вентиляционной функции легких из-за высокого стояния диафрагмы и значительного уменьшения объема легких.
Лечение
Лечение проводится с учетом этиологии П. При ревматизме, ревматоидном артрите, системной красной волчанке возникновение П. служит показанием для активной противовоспалительной терапии, включая глюкокортикостероидные гормоны в средних дозах (30—40 мг преднизолона в день). П. при периодической болезни купируют колхицином по 1 мг 2—3 раза в день с последующим переходом на поддерживающую дозу по 1 мг в день; глюкокортикостероидные гормоны в этом случае неэффективны. При адгезивном П. доза преднизолона должна быть меньшей — 15—20 мг в сутки; показана дыхательная гимнастика. При инфекционном П. назначают антибиотики (внутрь и внутриполостной а при туберкулезе проводят активную специфическую противотуберкулезную терапию в сочетании со средними и малыми дозами преднизолона.
При быстром накоплении экссудата независимо от этиологии П. производят пункцию серозной полости с удалением экссудата и последующим введением в полость гидрокортизона в дозе 50—100 мг, при необходимости применяют антибиотики. При сдавливающем перикардите показана перикардэктомия.
Профилактика состоит в раннем и адекватном лечении основного заболевания.
Библиография: Виноградова О. М. Периодическая болезнь, М., 1973; Насонова В. А. Системная красная волчанка, М., 1972; Нестеров А. И. Ревматизм, М., 1973; Оленева Т. Н. и Прохоров Е. П. Терапия антибактериальными препаратами и кортикостероидными гормонами больных туберкулезными плевритами, пневмоплевритами и полисерозитами, Пробл, туб., № 7, с. 24, 1960; С т р у к о в А. И. и Бегла р я н А. Г. Патологическая анатомия и патогенез коллагеновых болезней, М., 1963; С т р у к о в А. И. и С е-р о в В. В. Патологическая анатомия, М., 1979; Тареев E. М. Коллагенозы, М., 1965.
В. А. Насонова; М. Н. Ланцман (пат. ан.).
Периодическая болезнь и почечный амилоидоз у детей
Периодическая болезнь (синонимы: семейная средиземноморская лихорадка, доброкачественный пароксизмальный перитонит, рецидивирующий полисерозит, еврейская болезнь, армянская болезнь) — наследственное аутосомно-рецессивное заболевание, распространенное сред
Периодическая болезнь (синонимы: семейная средиземноморская лихорадка, доброкачественный пароксизмальный перитонит, рецидивирующий полисерозит, еврейская болезнь, армянская болезнь) — наследственное аутосомно-рецессивное заболевание, распространенное среди представителей древних народов Средиземноморья. Наиболее часто периодическая болезнь (ПБ) встречается у евреев-сефардов, армян, арабов, греков, турков, народов Кавказа и т. д., отсюда другие названия заболевания. Встречаемость ПБ среди евреев-сефардов по разным данным составляет от 1:250 до 1:2000 (частота носительства мутантного гена от 1:16 до 1:8), среди армян — от 1:100 до 1:1000 (частота носительства — от 1:7 до 1:4).
Среди 15 детей с ПБ, наблюдавшихся в Российской детской клинической больнице (РДКБ) в течение последних лет, 8 были армянами, 4 — дагестанцами, 1 — грек, 1 — с чеченскими и еврейскими корнями, 1 — русский.
Этиология и патогенез
В основе ПБ лежит точечная мутация в гене белка пирина, расположенного в коротком плече 16-й хромосомы (16q) рядом с генами аутосомно-доминантного поликистоза почек и туберозного склероза. Пирин — белок первичных гранул нейтрофилов, активно участвующий в регуляции процессов воспаления. Считается, что пирин стимулирует выработку противовоспалительных медиаторов, позволяет контролировать хемотаксис, стабилизирует мембрану гранулоцитов. Нарушение структуры этого белка, имеющее место при ПБ, приводит к повышению выработки провоспалительных медиаторов в лейкоцитах, активации микротубулярного аппарата и спонтанной дегрануляции первичных гранул лейкоцитов, активации молекул адгезии и усиленному хемотаксису лейкоцитов, результатом чего является воспаление.
На сегодняшний день известно 8 типов мутаций в С-концевом участке гена пирина, при которых происходит точечная аминокислотная замена. Наиболее распространены три мутации, на долю которых приходится более 90% случаев ПБ: М680I (замена изолейцина на метионин), М694V (замена валина на метионин), V726А (замена аланина на валин). Все три мутации насчитывают 2000–2500 лет, поэтому их иногда называют «библейскими», поэтому имеют преимущественное распространение у представителей древних народов, населявших земли вокруг Средиземного моря. Мутация М680I встречается в основном у армян, М694V и V726А — у всех этнических групп.
ПБ протекает в виде приступов, основой которых является спонтанная или спровоцированная дегрануляция нейтрофилов с выбросом медиаторов и развитием асептического воспаления преимущественно на серозных и синовиальных оболочках. В периферической крови повышается количество нейтрофилов и острофазовых белков (СРБ — С-реактивный реактивный белок, SAA — сывороточный белок амилоида А и др.). Раздражение медиаторами воспаления рецепторов приводит к развитию болевого синдрома, а воздействие большого количества эндогенных пирогенов на центр терморегуляции — к развитию лихорадки.
Клиническая картина и течение
Клинически ПБ проявляется возникающими через определенные интервалы (дни — недели — месяцы) стереотипными приступами лихорадки. Лихорадке могут сопутствовать болевые синдромы, связанные с развитием неспецифического воспаления в серозных и синовиальных покровах. В зависимости от пенетрантности генов эти синдромы могут быть изолированными или сочетаться, но каждый из них сохраняет свой ритм. Любая атака сопровождается лейкоцитозом, увеличением СОЭ и других воспалительных белков, повышением a- и b-фракции глобулинов, снижением активности миелопероксидазы нейтрофилов. Вне приступа дети чувствуют себя хорошо, лабораторные показатели постепенно нормализуются.
Лихорадка — наиболее частый и постоянный симптом при ПБ, встречается в 96–100% случаев. Особенностью лихорадки при ПБ является то, что она «не контролируется» антибиотиками и антипиретическими средствами. Изолированная лихорадка при ПБ, как правило, приводит к диагностическим ошибкам и расценивается как проявление ОРВИ.
Вторым по частоте симптомом ПБ является абдоминальный болевой синдром (асептический перитонит), который встречается в 91% случаев, а изолированно — в 55%. Клинически асептический перитонит мало отличается от септического со всем характерным для последнего симптомокомплексом: температура до 40°, резчайшая абдоминалгия, тошнота, рвота, угнетение перистальтики кишечника. Через несколько дней перитонит стихает, перистальтика восстанавливается. Подобная клиника часто является причиной диагностических ошибок, и больных оперируют по поводу острого аппендицита, перитонита, холецистита, кишечной непроходимости и др. Среди наблюдаемых нами детей 6 были ранее оперированы, причем 2 пациента — дважды: 4 — по поводу острого аппендицита, 2 — по поводу кишечной непроходимости, 1 — по поводу перитонита, 1 — острого холецистита. Как правило, в медицинской документации таких больных отмечается наличие «катарального аппендицита» и необходимость хирургического вмешательства не вызывает сомнений. Довольно типично то, что, со слов родителей, врачи, оперировавшие ребенка, в частных беседах отрицали действительное наличие аппендицита или перитонита.
Длительность лихорадочного и абдоминального вариантов ПБ обычно составляет от 1 до 3 дней, реже удлиняется до 1–2 нед.
Перитонит, как и суставной синдром, наиболее закономерен для детского возраста.
Суставной синдром характеризуется артралгиями, воспалением крупных суставов. Артриты и артралгии по различным данным наблюдаются в 35–80% случаев, причем в 17–30% они являются первыми признаками заболевания. В момент приступа появляются внезапные суставные боли в одном или нескольких суставах, которые могут сопровождаться отеком, гиперемией и гипертермией суставов. Продолжительность суставного варианта приступа ПБ составляет 4–7 дней, иногда удлиняясь до 1 мес. В отличие от изолированной лихорадки или пароксизмального перитонита при этом варианте ПБ артралгии часто сохраняются и после приступа, постепенно стихая в течение нескольких месяцев. Неспецифичность клинической картины при суставном варианте ПБ приводит к тому, что у больных диагностируют ревматоидный артрит, ревматизм, системную красную волчанку и пр. Отец одной из наших пациенток, по национальности армянин, длительные годы наблюдался с диагнозом «ревматоидный артрит», и только при выявлении ПБ у ребенка нами был генетически установлен тот же диагноз у него.
Торакальный вариант с плевральным синдромом встречается реже — около 40% случаев, изолированно — в 8%, в сочетании с абдоминальным синдромом — в 30%. При торакальном варианте развивается одно-двусторонний плеврит со стерильным выпотом. Длительность этого синдрома — 3–7 дней. Как правило, таким больным ошибочно устанавливается диагноз плеврита или плевропневмонии.
Кожные изменения во время приступа ПБ встречаются в 20–30% случаев. Наиболее типичной является рожеподобная сыпь, но могут встречаться пурпурные высыпания, везикулы, узелки, ангионевротические отеки. Иногда клинически ПБ протекает подобно аллергической реакции вплоть до отека Квинке и крапивницы.
Другими проявлениями ПБ могут быть головная боль, асептический менингит, перикардит, миалгия, гепатолиенальный синдром, острый орхит.
Среди наших пациентов у 12 ПБ протекала по абдоминальному варианту, у 3 — по абдоминально-суставному. 11 из них поступали в РДКБ с другими диагнозами: хронический холецистит, панкреатит, гастродуоденит, болезнь Крона, колит неясной этиологии, ревматоидный артрит, СКВ (системная красная волчанка), хронический гломерулонефрит и только 4 — с направляющим диагнозом «периодическая болезнь». Большинство больных поступают в отделение гастроэнтерологии с жалобами на рецидивирующие абдоминальные боли, при вовлечении почек с развитием протеинурии и нефротического синдрома — в отделение нефрологии, при немотивированной рецидивирующей лихорадке — в инфекционные и диагностические отделения.
Манифестация заболевания может приходиться на различный возраст. Описаны случаи довольно позднего манифестирования ПБ, после 20–25 лет. По нашим наблюдениям, у большинства больных первый приступ ПБ отмечался в возрасте 2–3 лет (9 пациентов), у 1 — с рождения, у 2 — в 0,5–1,5 года, у 2 — в 4–5 лет, у 1 — в 11–12 лет.
Частота и периодичность приступов варьируются у разных больных в широких пределах: от нескольких раз в неделю до 1–2 раз в несколько лет. У большинства больных приступы имеют довольно стабильный ритм. Однако в литературе описаны случаи, когда приступы могли прекратиться на несколько лет или, наоборот, возобновиться после длительного перерыва под воздействием внешних факторов (смена места жительства, женитьба или замужество, рождение ребенка, служба в армии и др.). У наших пациентов периодичность приступов была довольно постоянной: у 1-го — 2 раза в нед, у 4-х — 1 раз в нед, у 5-ти — 1 раз в 2–3 нед, у 2-х — 1 раз в мес, у 1 — 1 раз в 2–3 мес, у 1-го — 1 раз в 6–12 мес.
Через некоторое время от начала манифестации у большинства больных отмечается гепатомегалия, которая, по нашим наблюдениям, может варьироваться от +1 до +5 см. Постепенно развивается и спленомегалия, величина которой у некоторых пациентов достигала +7 см. Однако увеличение печени и селезенки выявляется не у всех больных. Очевидно, эти процессы зависят от частоты и количества перенесенных приступов и развития амилоидоза.
Амилоидоз как осложнение периодической болезни
Каждый приступ ПБ сопровождается выбросом большого количества медиаторов, образованием воспалительных белков. Из тканей и серозных покровов эти белки попадают в кровь, где длительно циркулируют. Таким образом, перед организмом стоит задача каким-то образом элиминировать эти белковые вещества. Чем чаще и выраженнее приступы ПБ, тем острее проблема утилизации. Одним из способов избавиться от избытка циркулирующих белковых молекул является их переработка с образованием нерастворимого белка — амилоида. Выражаясь образно, амилоид — это плотно упакованный белковый «мусор». Образование и отложение в тканях амилоида приводит к развитию амилоидоза.
Амилоидоз (от лат. amylum — крахмал) — собирательное понятие, включающее группу заболеваний, характеризующихся внеклеточным отложением белков в виде характерных амилоидных фибрилл. Эти нерастворимые фибриллярные белки могут быть локализованы в одном специфическом месте или могут быть распространены в различных органах, в том числе таких жизненно важных, как почки, печень, сердце и др. Такое накопление приводит к органной дисфункции, недостаточности органа и в конечном итоге смерти.
Структура амилоида идентична при всех его типах и представляет собой жесткие неразветвляющиеся фибриллы диаметром около 10 нм, обладающие складчатой β-кросс-конформацией, благодаря которой возникает эффект двойного лучепреломления в поляризованном свете при окраске Конго-красным. Окраска щелочным Конго-красным является наиболее распространенным и доступным методом выявления амилоида.
Амилоид состоит из фибриллярных белков (фибриллярный компонент, F-компонент) и гликопротеидов плазмы крови (плазменный компонент, P-компонент). Предшественники F-компонента различаются при различных видах амилоидоза (на сегодняшний день известно до 30 белков-предшественников, они определяют тип амилоидоза); предшественник Р-компонента один — сывороточный амилоидный Р-компонент (SAP), схожий с α-глобулином и СРБ.
Фибриллы амилоида и плазменные гликопротеиды образуют комплексные соединения с хондроитинсульфатами ткани с участием гематогенных добавок, среди которых основными являются фибрин и иммунные комплексы. Связи между белковыми и полисахаридными составляющими в амилоидном веществе особо прочные, что объясняет отсутствие эффекта при воздействии на амилоид различных ферментов организма, т. е. амилоид нерастворим.
При ПБ основой формирования фибриллярного компонента амилоида является сывороточный острофазовый белок SAA. SAA является a-глобулином, близким по своим функциональным свойствам к СРБ. SAA синтезируется клетками разных типов (нейтрофилами, фибробластами, гепатоцитами), его количество повышается во много раз при воспалительных процессах и опухолях. У человека выделено несколько типов SAA, и только фрагменты некоторых из них входят в состав амилоидных фибрилл, что, возможно, объясняет развитие амилоидоза только у части больных, несмотря на повышенную выработку SAA. Из сывороточного SAA-предшественника в тканях образуется АА-белок (белок амилоида А), который и является основой амилоидных фибрилл. Поэтому тип амилоидоза, развивающегося при ПБ, называется АА-амилоидоз.
Таким образом, основой развития амилоидоза при ПБ является избыточное образование белка-предшественника SAA. Но для образования амилоидного белка необходимы клетки, которые будут его синтезировать — амилоидобласты. Эту функцию выполняют в основном макрофаги-моноциты, а также плазматические клетки, фибробласты, ретикулоциты и эндотелиальные клетки. Макрофаги перерабатывают АА-белок в полноценные амилоидные фибриллы на своей поверхности и откладывают его в межуточной ткани. Поэтому наибольшее накопление амилоида при ПБ отмечается в органах, где макрофаги занимают фиксированное положение: почки, печень, селезенка. Постепенно все увеличивающиеся отложения амилоида приводят к сдавливанию и атрофии паренхиматозных клеток, склерозу и недостаточности органа.
Амилоидоз при ПБ болезни развивается по различным данным у 10–40% больных. Некоторые пациенты, несмотря на довольно частые приступы, не развивают амилоидоз вовсе. Вероятно, развитие амилоидоза зависит от особенностей строения белка-предшественника у данного пациента и генетической способности макрофагов синтезировать амилоид.
Несмотря на то, что амилоидоз может развиваться в любом органе и ткани, амилоидное поражение почек играет определяющую роль для прогноза и жизни больного ПБ. При развитии АА-амилоидоза почки поражаются в 100% случаев.
В почках роль амилоидобластов выполняют мезангиальные и эндотелиальные клетки.
В процессе отложения амилоида в почечной ткани и вызванного им поражения органа можно проследить определенную стадийность. Выделяют 4 стадии амилоидоза почек: латентную (диспротеинемическую), протеинурическую, нефротическую (отечную) и уремическую (азотемическую).
В латентную стадию изменения в почках незначительны. Отмечаются нарушения гломерулярного фильтра в виде очагового утолщения, двухконтурности мембраны и аневризм ряда капилляров. В гломерулах амилоида нет или он обнаруживается не более, чем в 25% клубочков.
Ведущим в патогенезе этой стадии амилоидоза является значительный синтез и повышение в плазме крови концентрации белков-предшественников амилоидоза, т. е. диспротеинемия. Клинически у детей на фоне приступов ПБ может развиваться гипохромная железодефицитная анемия, гиперпротеинемия, диспротеинемия с увеличением глобулинов α2, β и γ, отмечается высокое содержание фибриногена и сиалопротеинов. Характерны увеличение и уплотнение печени и селезенки.
Изменения в моче поначалу отсутствуют или носят транзиторный характер, однако со временем протеинурия становится постоянной и более выраженной, часто наблюдается микрогематурия и цилиндрурия. Появление постоянной протеинурии характеризует переход во вторую, протеинурическую, стадию.
В протеинурической стадии амилоид появляется не только в пирамидах, но и в половине клубочков почек в виде небольших отложений в мезангии, отдельных капиллярных петлях, а также артериолах. Отмечается выраженный склероз и амилоидоз стромы, сосудов, пирамид и интермедиарной зоны, что приводит к атрофии многих глубокорасположенных нефронов.
Продолжительность этой стадии, как и предыдущей, колеблется от нескольких месяцев до многих лет. По мере нарастания тяжести амилоидоза усугубляются лабораторные показатели выраженной активности процесса: значительная протеинурия и диспротеинемия, гиперфибриногенемия, СРБ, гиперкоагуляция. Дальнейшее отложение амилоида в почечной ткани и нарастающая протеинурия приводят к развитию отечного синдрома, появление которого свидетельствует о переходе заболевания в третью, отечную, стадию.
В отечную (нефротическую) стадию амилоидоза количество амилоида в почках увеличивается. Пораженными оказываются более чем 75% гломерул. Прогрессирует склероз интерстиция и сосудов, в пирамидах и интрамедиарной зоне склероз и амилоидоз имеют выраженный диффузный характер.
Клинически эта стадия амилоидоза представлена полным нефротическим синдромом, хотя иногда может наблюдаться неполный (безотечный) нефротический синдром. Протеинурия становится массивной и, как правило, неселективной; нарастают циллиндры. Гематурия бывает редко и, как правило, незначительна. Нарастают гепатоспленомегалия, гипопротеинемия, усиливаются диспротеинемия с дальнейшим повышением уровня α1-, α2-, и γ-глобулинов, гиперфибриногенемия, гиперлипемия. Со временем появляется артериальная гипертензия, нарастает азотемия, прогрессирует почечная недостаточность.
Уремическая (азотемическая) стадия развивается в финале заболевания. В связи с нарастающим амилоидозом и склерозом наблюдаются гибель большинства нефронов, их замещение соединительной тканью, развивается ХПН (хроническая почечная недостаточность).
Клиническими особенностями ХПН при амилоидозе, отличающими ее от ХПН вследствие других заболеваний, является сохранение нефротического синдрома с массивной протеинурией, часто определяются большие размеры почек, характерно развитие гипотензии.
Часто выражен ДВС-синдром (синдром диссеминированного внутрисосудистого свертывания крови) в виде пурпуры, носовых, желудочных и кишечных кровотечений. Возможны тромбозы почечных сосудов с развитием инфарктов ишемического или геморрагического типа.
Мы наблюдали развитие амилоидоза у 4-х детей с ПБ (26% наблюдаемых больных). Транзиторная протеинурия у них появлялась на 7–8 год от манифестации заболевания, через 2–3 года она принимала постоянный характер. У 2-х детей через 1,5–2 года после установления постоянной протеинурии развился нефротический синдром, который у одного ребенка перерос в ХПН.
С момента развития нефротического синдрома детям диагностировали хронический гломерулонефрит и назначали соответствующее лечение глюкокортикоидами, которое не имело эффекта. В дальнейшем заболевание расценивали как СКВ и гормонорезистентый вариант гломерулонефрита, дети получали терапию цитостатиками, также без эффекта. Диагноз «периодическая болезнь, амилоидоз почек» в обоих случаях был впервые установлен в РДКБ.
Развитие амилоидоза в определенной степени зависит от количества перенесенных ребенком приступов ПБ. Среди наших пациентов амилоидоз почек выявлялся у тех, кто перенес более 130–150 приступов, тогда как у детей с меньшим количеством приступов признаков амилоидоза и поражения почек не отмечалось. Причем дети с нефротическим синдромом перенесли наибольшее число приступов — около 240 и 260. Следует отметить, что подобная закономерность не является абсолютной и амилоидоз может развиться и при меньшем количестве приступов ПБ.
Диагностика периодической болезни и амилоидоза
При типичном течении периодической болезни ее диагностика не представляет трудностей. Наибольшая проблема заключается в незнании большинством врачей этой патологии, что приводит к плохой выявляемости даже при наличии симптомов.
Диагностика ПБ основывается на 5 пунктах.
Диагностика АА-амилоидоза представляет значительную трудность. В большинстве случаев АА-амилоидоз своевременно не диагностируется, даже когда имеются клинические признаки заболевания. Причиной этого являются, с одной стороны, неспецифичность симптомов заболевания, а с другой — отсутствие у большинства врачей настороженности в отношении амилоидоза, что связано в том числе с его малой распространенностью у детей. Однако наши представления о частоте амилоидоза у детей ошибочны, и выявляемые случаи представляют собой лишь «вершину айсберга». Как показывают последние исследования, проведенные у взрослых пациентов, амилоидоз при жизни не диагностирован у 83% больных.
При постановке диагноза ПБ в большинстве случаев у врача возникает настороженность в отношении амилоидоза. Но часто первые подозрения на АА-амилоидоз могут возникнуть у педиатра при лечении больных с нефротическим синдромом, резистентных к стандартной глюкокортикоидной терапии.
Только изучение материалов биопсии с обязательным окрашиванием Конго-красным и поляризационной микроскопией позволяет поставить окончательный диагноз АА-амилоидоза. Помимо этого для диагностики можно использовать специфические антитела к АА-фибриллам. Наиболее достоверной является биопсия почки. Частота выявления АА-амилоидоза в этом случае достигает 90–100%. Чем более распространен процесс, тем больше вероятность выявления АА-амилоида в других местах (желудочно-кишечный тракт (ЖКТ) — слизистая и подслизистая, слизистая десны, прямая кишка, жировая биопсия). Наиболее информативной среди непочечных биопсий является биопсия стенки ЖКТ и прямой кишки, при которой вероятность выявления амилоида составляет 50–70%.
Лечение
При периодической болезни основой терапии является назначение колхицина. Колхицин обладает антимитотическим эффектом в отношении амилоидобластов при периодической болезни — макрофагов и стабилизирует мембрану нейтрофилов, препятствуя выбросу пирина. Колхицин назначается пожизненно в дозе 1–2 мг/сут. Он хорошо переносится, иногда возникают диспептические явления, которые не требуют полной отмены препарата. Колхицин в большинстве случаев полностью предотвращает появление приступов ПБ или значительно снижает их частоту и выраженность, предотвращает развитие амилоидоза почек, снижает выраженность его проявлений. При почечной недостаточности дозу снижают исходя из степени снижения клубочковой фильтрации. Препарат может быть временно отменен при острых инфекциях у ребенка.
Нами наблюдался мальчик, который был направлен в РДКБ с генетически установленным диагнозом периодической болезни в возрасте 16 лет. Приступы ПБ отмечались у него с 4-летнего возраста, протекали с периодичностью 1 раз в 2–3 нед в виде лихорадки с абдоминальным болевым синдромом, 1–2-кратной рвотой, головной болью и выраженной слабостью. Приступы длились около суток, затем в течение 1–2 дней была столь выраженная слабость, что мальчик не мог встать с постели, не посещал школу. Признаков амилоидоза не выявлялось.
В РДКБ ребенку был назначен колхицин в дозе 2 мг/сут. За последующие 2 года наблюдения число приступов резко снизилось до 1–2 раз в год, а в течение последних 10 месяцев не было ни одного. Сейчас юноша успешно обучается в вузе, живет в общежитии в другом городе, чувствует себя хорошо.
В лечении ПБ и профилактике амилоидоза необходима организация правильного питания ребенка. Увеличение общего количества белка в рационе стимулирует амилоидогенез, тогда как белок печени и мышцы сердца ингибируют его. Рекомендуется диета со сниженным на 50% содержанием животного (особенно казеина) и растительного белков и увеличением продуктов, содержащих крахмал. Диета должна быть достаточно обогащенной фруктами, овощами и другими шлакогонными продуктами. Белок предпочтительнее давать ежедневно (100 г печени, сырой или кулинарно обработанной). Печень употребляют годами, в виде повторных многомесячных курсов. Используют гепатотропные препараты повторными курсами: по 2–4 мес Эссенциале, Липоевую кислоту.
Прогноз
При своевременной постановке диагноза и назначении колхицина прогноз ПБ благоприятен.
При отсутствии терапии наибольшую опасность представляет развитие почечного амилоидоза, который, по сути, является единственной причиной смерти больных с ПБ. Анализ заболеваемости у взрослых и детей показывает, что при естественном течении периодической болезни приблизительно у 50% больных терминальная стадия почечной недостаточности развивается через 5 лет от момента появления протеинурии, у 75% — в течение 10 лет.
По вопросам литературы обращайтесь в редакцию.
А. В. Малкоч, кандидат медицинских наук
РГМУ, Москва