Множественные врожденные пороки развития (МВПР)
О множественных врожденных пороках развития (МВПР) говорят тогда, когда отмечается нарушение структуры и функционирования минимум двух органов или систем. Обычно такие изменения сильно отражаются на жизнедеятельности, нередко становятся причиной внутриутробной гибели плода или неонатальной смерти. Некоторые множественные врожденные пороки развития (МВПР) не сокращают продолжительность жизни, но снижают ее качество, требуют постоянного контроля со стороны специалистов или определенных условий содержания.
Причины множественных врожденных пороков развития
Все этиологические факторы МВПР можно разделить на две группы:
Эти причины приводят к хромосомным, генным и геномным нарушениям, влияют на процессы клеточного и тканевого развития. Большое число множественных врожденных пороков (МВПР) отличаются типом наследования, патогенезом, клиническими проявлениями, прогнозом для пациента.
Примеры множественных врожденных пороков развития (МВПР)
Другие виды множественных врожденных пороков развития (МВПР) оказывают существенное влияние на жизнедеятельность человека. При этом лечение возможно только симптоматическое, так как изменить дефекты в генах или хромосомах невозможно.
Диагностика множественных врожденных пороков развития (МВПР) может осуществляться дородовыми и послеродовыми способами. В первом случае проводятся скрининговые исследования, которые включают в себя УЗИ плода и анализ крови на специфические белки. Такое сочетание позволяет выявить большое количество множественных врожденных пороков развития (МВПР). Врач также может назначать дополнительные методы диагностики: биопсию ворсин хориона и амниоцентез. В этом случае получают не кровь матери, а материал плода, который затем отправляют на генетическое исследование.
Послеродовые методики также подразумевают генетические тесты, которые позволяют выявлять мутации в генах или определять кариотип. Пройти такое исследование можно в медико-генетическом центре «Геномед».
Подозрение на синдромальную патологию что это множественные пороки развития
В соответствии с вышеприведенной классификацией в этом разделе будут рассмотрены клинические особенности синдромов с множественными врожденными аномалиями, при наследственных дефектах обмена, факоматозах, неврологических и нервно-мышечных заболеваниях.
Синдромы с множественными врожденными аномалиями
Понятие «врожденные аномалии» довольно широкое: оно включает как врожденные пороки, так и отклонения в строении тела, представляющие чаще всего крайние варианты нормы (эпикант, большие уши и т. д.). Последние называют «малыми аномалиями». Если этих аномалий много, то они формируют специфический диспластичный облик.
Патогенез интеллектуального недоразвития при этих нарушениях в большинстве случаев остается неясным, но он может быть принципиально различным в разных группах заболеваний.
В некоторых случаях в искаженное формирование мягких тканей и скелета по вероятностным закономерностям вовлекается и ЦНС, т. е. аномалия структуры мозга является одним из симптомов всего комплекса множественных врожденных аномалий развития. В таких случаях порок развития мозга является факультативным симптомом того или иного синдрома. Этот дефект формирования мозга может быть анатомически явным (микроцефалия и др.), а может и проявляться негрубыми нарушениями структуры мозговой ткани, например гетеротопией нейронов в отдельных участках коры, нарушениями пропорций нейронов и глии и т. п.
Возможен и другой механизм повышенного сочетания умственной отсталости с тем или иным синдромом. При некоторых из этих заболеваний интеллектуальный дефект может носить вторичный по отношению к основной симптоматике синдрома характер. В этих случаях искаженное формирование органов и тканей, вызывая резкое нарушение каких-либо функций организма, может сказаться на развитии мозга уже в постнатальном периоде. Та или иная нарушенная функция организма, изменение которой вызвано внутриутробным дисгенезом, может обусловить повреждение или замедление созревания анатомо-физиологических структур мозга.
Во многих же случаях синдромы с множественными аномалиями развития представляют собой по сути дела еще не раскрытые наследственные дефекты обмена с началом во внутриутробном периоде, но с продолжающимся постнатально действием патогенного фактора. Они сопровождаются биохимическими нарушениями в клетках мозга в течение всего периода развития организма. При таких поражениях умственная отсталость является постоянным симптомом, а имеющаяся «диспластичность» больного еще не свидетельствует о поражении, законченном во внутриутробном периоде.
Наследственные заболевания, входящие в категорию синдромов с множественными врожденными аномалиями, представлены тремя этиологически различными группами: 1) хромосомные заболевания; 2) генетические синдромы с неясным типом наследования; 3) моногенно наследуемые синдромы.
Значительная часть синдромов с врожденными аномалиями остается еще этиологически неясной и клинически неклассифицированной, являясь источником дальнейшей дифференциации новых нозологических форм.
Подозрение на синдромальную патологию что это множественные пороки развития
Сочетание множественных врожденных пороков развития пищеварительной системы, сердечно-сосудистой системы представляет клинический интерес с точки зрения сложности прижизненной диагностики у новорожденных. В связи с редкостью патологии мы бы хотели представить в данной статье клинический случай из практики.
Целью нашего исследования является изучение клинико-лабораторных особенностей течения множественных пороков развития у новорожденных.
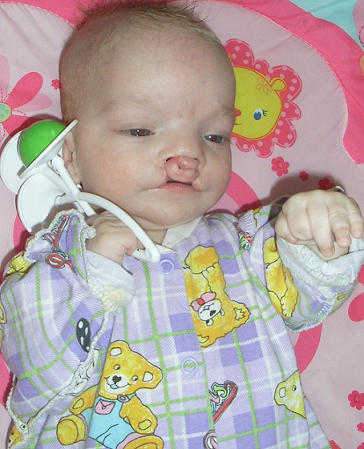
Материалы и методы. Описанный клинический случай сочетанных множественных пороков развития, ребенка Г., в возрасте 3 месяцев находящегося в ОРИТ КГП «ОДКБ». Данному ребенку были проведены комплексное обследование, в том числе R-графия ОГК, НСГ. (Рис.1)
При поступлении жалобы на: пенистое отделяемое изо рта с первого дня жизни.
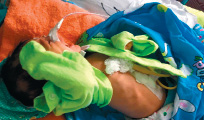
Объективные данные при поступлении в ОРИТ: Состояние ребенка очень тяжелое. Тяжесть обусловлена ДН, поражением ЦНС, интоксикацией, на фоне незрелости, врожденного порока развития ЖКТ. Доставлен на спонтанном дыхании. В сознании, на осмотр реагирует повышением двигательной активности, болезненная гримаса на лице, подстанывает. Установлен зонд в пищевод, около 10см, выделяется вязкий слизистый секрет. Признаки незрелости. Ребенок пониженного питания. Тепло удерживает плохо.
26.09.16 г. выполнено оперативное вмешательство: проведена верхнесрединная лапаратомия. Ревизия верхних отделов ЖКТ с мобилизацией дистального отдела пищевода через пищеводное отверстие диафрагмы до места сообщения с трахеей. Последний у выхода из трахеи прошит, перевязан, иссечен. Культя пищевода так же прошита, перевязана. Произведена гастростомия с выведением через отдельный разрез в левом подреберье катетера фолея №2. Диагноз после операции: МВПР, ВПР ЖКТ. Атрезия пищевода с нижним трахеопищеводным свищом.
Находился на ИВЛ в течение 1.5 месяца, учитывая эффективное самостоятельное дыхание ребенок переведен на спонтанное дыхание. 24.11.16 состояние ребенка стабилизировано, переведен в х/о, выписан с улучшением.
Проведенное исследование за наблюдаемый период:
Группа крови А(II) вторая Rh(+)положительный.
ОАМ от 25.09.16: с/ж,проз., белок 0,05 г/л., лейкоциты 2-3, эритроциты Ед,эпителий 3-4.
ОАМ от 14.11. с\жёлтая, белок 0,058‰, эритроциты 1-2, ураты ++++, дрожжевые грибки +.
Анализ ликвора (11.10.16): б/ц, прозр., цитоз – 1,0, белок – 1,0/л, сахар – 1,2ммоль/л.
Коагулограмма от 27.09.16г: ПТИ-82, АЧТВ-56, РФМК-отр
Коагулограмма от 14.11. МНО 1,56,Фибриноген 0,94, ПТИ 59%, АЧТВ 43.7, ПТВ 17.9, ТВ 13.3.
Копроскопия от 24.09.16г:консистенция-каш, цвет-зел,слизь+,лейкоциты 4-6,бактерии+, нейт.жир++, дрож.грибки+.
Копроскопия от 06.12.16г.: неоф., желт., нейтр. жир++, бактерии ++++, слизь +, лейк. 1-0 в п/зр. Простейшие и я/гл. не обнаруж
Бак.посев кала от 26.09.16г: отр.
Кровь на стерильность от 19.10.16 не стерильно. Staphylococcus saprophyticus 103/
Кровь на стерильность и чувствительность к антибиотикам от 29.11.16: стерильно
Кровь на ВИЧ от 05.12.16г.: № 4210 отрицат.
ПЦР от 30.09.16г: гепатит В-отр, гепатит С-отр
Инструментальные методы исследования:
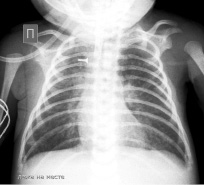
Рентген грудной клетки от 25.09.16- Атрезия пищевода с нижним трахеопищеводным свищом. (Рис.№2)
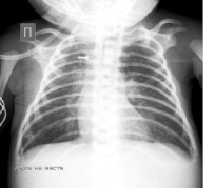
Рентген грудной клетки от 04.10.16 – R-приз. Аспирационной пневмонии.
ЭКГ от 26.09.16: Синусовая тахикардия.Нагрузка на правое предсердие. Неполная блокада правый ножки пучка Гиса.Выраженные метаболические изменение.
НСГ от 25.09.16 –Перивентрикулярное кровоизлияние 2 степени с обеих сторон. Гипоксическое повреждение. Множественные псевдокисты сосудистых сплетений. ВУИ?
НСГ от 28.09.16- Перивентрикулярное кровоизлияние 2 степени с обеих сторон. Гипоксическое повреждение. Множественные псевдокисты сосудистых сплетений
НСГ от 22.11.16. Умеренная вентрикулодилятация. Псевдокиста правого сосудистого сплетения в стадии лизиса..
УЗИ от 15.11.16. УЗИ картина гепатомегалии. ССЖ.Реактивный панкеатит. УЗИ картина мочекислого диатеза.
ЭХО-КС от 26.09.16: Дефект межпредсердный перегородки. Расширение легочной артерии.
Окулист от 17.11.16 Ангиопатия сетчатки.
Невропатолог от 18.11.16 последствия энцефалопатии смешанного генеза (гипоксически-геморрагически-инфекционного) тяжелой степени тяжести, синдром двигательных нарушений, синдром повышенной нейрорефлекторной возбудимости.
Консультирован сурдологом 02.12.16г.: периферический слух в норме. Повторить скрининг в 3,6,9,12 мес.
Осмотрен ЛОР врачом 06.12.16г.: Со стороны ЛОР органов без особенностей.
Проведено лечение в ОРИТ.
— Выхаживание в кувезе;
— Дробное энтеральное питание, под контролем усвоения.
— Обезболивание в послеоперационном периоде – анальгин+димедрол по 0,1мл;
— С целью седации – брюзепам в послеоперационном периоде (17.02.16 – 19.02.16), фенобарбитал 7мг\кг\сут с 28.09.16;
С противогрибковой целью Нофлук 16 мг х 1 р №20
— ИТ в режиме парентерального питания глюкозо-солевыми смесями + Аминоплазмаль+ Липофундин №15;
— с заместительной целью введен Ig G поливалентный человеческий –препарат «Октагам» в дозе 2,5 гр.№2
— Сердечные гликозиды по 0,1мл*2р №3:
— С гемостатической целью – этамзилат 0,5мл*4р + амри К-0,2 мл*3 дня;;
-Сурфактантная терапия – «Куросурф» 240мг(по 120мг в каждый бронх) №1;
— С кардиотонической целью – дофамин 5мкг\кг\мин №7
С целью возмещения дефицита факторов коагуляционного гемостаза – СЗП: л/ф, 100мл №1;
С целью возмещения анемии: эритровзвесь А(II) Rh(+), 50мл №3
С целью возмещения тромбоцитов крови вводился тромбоконцентрат №8
На фоне проведенной терапии состояние ребенка с положительной динамикой.
По зонду из пищевода – слизистое отделяемое, по гастростоме отделяемого нет. Живот поддут, мягкий. Кормится по 60мл дробно через гастростому. Питание усваивает. в весе прибавляет. Стал кратковременно фиксировать взгляд, кратковременно следить. Стул регулярный, кашицеобразный. Мочится свободно. Мама обучена уходу за ребёнком: кормлению через гастростому, установке зонда в пищевод.
Основной задачей при данной патологии — предотвратить заброс содержимого пищевода в дыхательные пути. Прогнозирование исходов лечения атрезии пищевода должно основываться на степени недоношенности, наличии сопутствующих аномалий развития, в том числе, пороков сердца, необходимости перевода новорожденного после рождения на искусственную вентиляцию легких, протяженности диастаза между отрезками пищевода.
Запланировано постепенное увеличение разового объема кормления до физиологической потребности. На фоне проводимого лечения состояние несколько стабилизировалось. В динамике запланирован второй этап оперативного лечения с целью удаления гастростомы.
Выводы. Несмотря на отсутствие пренатальной диагностики, своевременная постнатальная диагностика и проведение оперативного лечения у новорожденного с МВПР (ВПРЖКТ и ВПС) на фоне недоношенности и осложнений был, достигнут благоприятный исход в нашем случае. В связи с этим представили данный клинический случай и алгоритм диагностики и лечения.
Самые частые патологии плода во время беременности. Обзор синдромов
Данный вид исследований позволяет обнаружить более 99 % плодов с синдромом Дауна (или трисомия по 21 хромосоме), синдромом Эдвардса (трисомия по 18 хромосоме),синдромом Патау (трисомия по 13 хромосоме). Хромосомные аномалии имеют место в 1 из 150 случаев деторождения. В случае наличия у плода болезни родители при помощи врача-генетика тщательно взвешивают возможности современной медицины и свои собственные в плане реабилитации ребёнка. В результате семья принимает решение о судьбе данного ребёнка и решает вопрос о продолжении вынашивания
Пренатальная диагностика подразделяется на инвазивную и неинвазивную.
Какие же основные синдромы выявляет НИПТ
Хромосомное заболевание характеризуется комплексом множественных пороков развития и трисомией 18 хромосомы. Описан в 1960 году Джоном Эдвардсом. Популяционная частота по миру 1:5000 на 2016 г. Дети с трисомией 18 чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии 21 хромосомы. Основные признаки синдрома Эдвардса: малый вес, аномалии форм черепа, пороки сердца, дефекты межжелудочковой перегородки, умственная отсталость.
Частота – 1:16000 родов. Характерны нарушения развития нервной системы, глаз, выраженная деформация лица, пороки развития сердца и других внутренних органов. Характерна глубокая задержка умственного развития. 90% детей умирают в возрасте до 1 года.
Гоносомные анеуплоидии:
Если в клетке присутствует только одна хромосома вместо двух, это называется моносомией. Частота встречаемости – 1:2500. Возникает в случае отсутствия второй половой хромосомы, встречается только у девочек. Единственная моносомия, при которой возможно рождение живого ребенка. Хромосомная болезнь, сопровождающаяся характерными аномалиями физического развития, низкорослостью и половым инфантилизмом.
Частота – 1:500. Когда в клетках с мужским кариотипом присутствует дополнительная Х-хромосома. Встречается только у мальчиков. Это заболевание, при котором особи мужского пола имеют дополнительную Х-хромосому. Обычно женщины имеют пару ХХ хромосом, а мужчины пару ХY хромосом, однако при этом заболевании мужчины имеют по крайней мере две Х-хромосомы и хотя бы одну Y хромосому. Синдром Клайнфельтера является не только самой частой формой мужского гипогонадизма, бесплодия, эректильной дисфункции, но и одной из наиболее распространённых эндокринных патологий, занимая третье место после сахарного диабета и заболеваний щитовидной железы.
Частота – 1:1000. В клетках присутствует дополнительная Y-хромосома. Встречается только у мальчиков. Носитель синдрома имеет дополнительную Y-хромосому, общий хромосомный набор составляет 44 аутосомы и три половые хромосомы. Наличие второй Y-хромосомы в большинстве случаев не ведёт к каким-либо физическим отклонениям. В то же время, многие мужчины с XYY-синдромом имеют одну или несколько особенностей. При рождении они имеют нормальный рост, но часто быстрее растут в детстве. В среднем, во взрослом состоянии носитель выше, чем 75% мужчин того же возраста. Некоторые мужчины с синдромом XYY имеют небольшие нарушения координации движений, в результате чего могут казаться неуклюжими. Фертильность чаще всего не нарушена, обычно такие мужчины гетеросексуальны и имеют нормальную сексуальную функцию. Тем не менее, описаны случаи существенного снижения фертильности, вплоть до бесплодия. У небольшого числа носителей также повышен уровень половых гормонов, связанных со сперматогенезом, что может вести к бесплодию ввиду нарушения образования спермы. Неизвестно, насколько высоко число случаев бесплодия у мужчин с XYY-синдромом. IQ находится в пределах нормы, но часто несколько ниже, чем у родных братьев и сестёр. Примерно половина носителей имеет проблемы с обучением, в частности, могут быть нарушения и чтения. Может быть повышен риск поведенческих проблем, таких как синдром гиперактивности, мужчины с XYY-синдромом часто импульсивны и эмоционально незрелы.
Частота – 1:1000. В клетках присутствует 3 копии Х-хромосомы. Встречается только у девочек.
В большинстве случаев носители дополнительной X-хромосомы — женщины без заметных признаков патологии, поэтому при медицинских исследованиях 90% трисомиков по X-хромосоме остаются не выявленными. Развитие может протекать с некоторыми нарушениями, могут возникнуть проблемы с координацией, моторикой и развитием речи. В некоторых случаях отмечен меньший размер головы (без заметного снижения умственных способностей). Трисомия по X-хромосоме не приводит к значительным нарушениям фертильности, в большинстве случаев может проявляться только в более ранней менструации.
Врожденные аномалии развития

Пороки развития представляют собой стойкие морфологические изменения органа или организма в целом, выходящие за пределы вариаций нормы и возникающие внутриутробно в результате нарушения развития зародыша либо плода, иногда – после рождения ребенка вследствие нарушения дальнейшего формирования органов. Эти изменения вызывают нарушения соответствующих функций. Синонимами термина “пороки развития” являются “врожденные пороки”, “аномалии развития”, “дисплазии”. Однако под аномалиями развития и дисплазиями понимают только такие пороки, при которых анатомические изменения не приводят к существенному нарушению функций, например деформации ушных раковин, не обезображивающие лица больного и существенно не отражающиеся на восприятии звуков. Грубые пороки развития, при которых обезображивается внешний облик ребенка, нередко называют уродствами. Однако термин “уродство” – понятие скорее социальное, чем медицинское.
ПРИЧИНЫ. ПОРОКИ И ИХ ВИДЫ
Причины возникновения врожденных пороков вообще и нервной системы в частности весьма разнообразны. Их могут обусловливать мутации, а также их сочетанное воздействие. Г. И. Лазюк (1982 г.) выделяет следующие причины врожденных пороков:
1) эндогенные (внутренние) факторы:
а) изменения наследственных структур (мутации);
б) “перезревание” половых клеток;
в) эндокринные заболевания;
г) влияние возраста родителей;
2) экзогенные (внешние) факторы:
а) физические – радиационные, механические воздействия;
б) химические – лекарственные препараты, химические вещества, применяемые в промышленности и в быту, гипоксия, неполноценное питание, нарушения метаболизма;
в) биологические – вирусные заболевания, протозойные инвазии, изоиммунизация.
Одной из главных причин пороков развития являются мутации. В организме они происходят постоянно (спонтанные мутации) под воздействием естественного фона радиации и процессов тканевого метаболизма. При дополнительном воздействии на организм ионизирующего излучения или химических мутагенов происходят индуцированные мутации. Мутации могут быть генными, хромосомными и геномными. Первые представляют собой новые молекулярные состояния гена. С мутацией единичных генов связано около 13% пороков. Хромосомные мутации – это изменения хромосом в виде транслокации, делеции, дупликации и инверсии. Геномные мутации – изменение числа хромосом или хромосомных наборов. Хромосомные и геномные мутации индуцируют развитие хромосомных болезней. Под “перезреванием” половых клеток понимают комплекс изменений в яйцеклетках и сперматозоидах, произошедших от момента их полного созревания до образования зиготы. Они наблюдаются в основном при увеличении времени от эякуляции до слияния спермия с яйцеклеткой и связаны преимущественно с изменением рН среды в половых путях, снижением подвижности сперматозоидов, с нарушением проходимости труб. Следствием “перезревания”, по-видимому, является нерасхождение хромосом, что в дальнейшем проявляется геномными мутациями.
Среди эндокринных заболеваний, вызывающих пороки развития, главную роль играет сахарный диабет. Пороки развития у детей возникают как при клинически проявляющихся, так и при латентных формах заболевания у матери, но особенно часто – у женщин, которые заболели в препубертатном периоде. Зависимость состояния ребенка от возраста родителей, в котором произошло его зачатие, хорошо известна. Так, у женщин старше 35 лет и мужчин старше 40 лет значительно увеличивается риск рождения ребенка с хромосомными болезнями, обусловленными числовыми изменениями хромосом. У отцов с возрастом повышается риск рождения ребенка с пороками, обусловленными вновь возникшими доминантными мутациями. Тератогенный эффект может наступить при воздействии ряда ионизирующих излучений и зависит от вида и энергии радиоизотопов, длительности их воздействия (острое облучение опаснее хронического) и суммарной дозы, а также от срока беременности (чем меньше, тем больше радиочувствительность плода) и индивидуальной чувствительности. Поглощенная плодом доза излучения в 10 рад в первую и в 20 рад – во вторую половину беременности может вызвать изменение его развития, в первую очередь увеличение патологии со стороны ЦНС (микроцефалия, нарушение миелинизации, катаракта), недостаточность эндокринной и иммунной систем. Тератогенная роль механических факторов (давление матки на плод при маловодии, шум, вибрация и др.) в развитии пороков центральной нервной системы пока еще окончательно не выяснена. Амниотические тяжи, особенно амниотические сращения, приводят к развитию амниотических перетяжек на конечностях, колобоме лица. Исследования тератогенного действия химических веществ, в том числе и медикаментов, особенно интенсивно стали проводиться с 1961 г., когда установили, что в результате приема женщинами седативного препарата талидомида в начале беременности дети рождаются с синдромом талидомидной эмбриопатии, проявляющимся в основном агенезией или гипогенезией длинных трубчатых костей, иногда – пороками развития глаз, ушей, сердца, почек, половых органов.
Из огромного количества медикаментов, тератогенный эффект которых доказан в эксперименте, на человека тератогенно действуют лишь определенные противосудорожные препараты (фенитоин, фенобарбитал), антикоагулянты (варфарин), противоопухолевые средства (миелосан, эндоксан) и антимиотические (колхицин) средства, антиметаболиты (аминоптерин). Антибиотики, принимаемые беременной, могут оказывать патологическое влияние на развитие плода. Однако истинных пороков развития они не вызывают. Особого внимания заслуживает внутриутробное повреждение плода в результате хронического употребления алкоголя в течение беременности. Еще в 1959 г. Л. А. Богданович отмечала, что у женщин, хронически употребляющих спиртные напитки, дети в 34,5% случаев рождаются недоношенными, в 19% – физически ослабленными, в 3% случаев – с выраженными пороками развития. Позже был описан синдром алкогольных эмбриофетопатий. Для него характерны врожденная гипоплазия и постнатальный дефицит роста и массы тела, общая задержка физического и психического развития, микроцефалия, короткие и узкие глазные щели, узкий скошенный лоб, эпикант, узкая красная кайма верхней губы, гипоплазия нижней челюсти. Он часто сопровождается гиперрефлексией, тремором, изменчивым мышечным тонусом, реже – спонтанными клоническими судорогами, опистотонусом, слабостью сосательного рефлекса. Кроме того, могут развиваться пороки сердца, почек, половых органов, конечностей. Установлено, что в первые годы жизни у таких детей сохраняется отставание в психомоторном, прежде всего речевом, развитии, часто сочетающееся с гипервозбудимостью и двигательной расторможенностью. Специфической особенностью интеллектуальных нарушений у этих детей служит наличие нерезко выраженной интеллектуальной недостаточности и эмоционально-личностной незрелости. Имеют место также отдельные признаки “лобной психики”, что проявляется малой критичностью, эйфорией, импульсивностью, слабой регуляцией произвольной деятельности.
Непосредственно гипоксия крайне редко является причиной пороков. Гипоксия может лишь индуцировать развитие пороков мультифакториального происхождения, например гидроцефалию. По-видимому, чаще пороки вызывают местное нарушение кровообращения, связанное с окклюзией сосудов. Неполноценное питание как тератогенный фактор действует при дефиците микроэлементов, особенно цинка, что обычно наблюдается в случаях хронических энтероколитов, безмясной диеты, приема больших доз салицилатов. Это может индуцировать пороки развития преимущественно ЦНС – главным образом гидроцефалию, микрофтальмию или анафтальмию, иногда – искривления позвоночника, расщелины неба, пороки сердца, грыжи.
Из биологических факторов наибольшее значение в развитии пороков имеют вирусы краснухи и цитомегалии. При заболевании краснухой (даже в скрытой форме) в I триместре беременности в 20-22% случаев развивается эмбриопатия. У новорожденных она проявляется субтотальной катарактой, микрофтальмией, реже – пороками сердца и глухотой, обусловленной поражением полукружных каналов. У части таких детей наблюдается микроцефалия, иногда – гидроцефалия. У детей, инфицированных цитомегаловирусом, возможно любое из приводимых ниже клинических состояний: низкая масса при рождении, гепатоспленомегалия, гепатит и желтуха новорожденных, тромбоцитопения, микроцефалия, хориоретинит, паховая грыжа, атрезия желчных протоков, поликистоз почек. Цитомегаловирус также поражает внутреннее ухо, приводя к глухоте. Вирус также может поражать зубы, вызывая аномалии прикуса, желтый цвет эмали зубов. Новорожденный может быть заражен цитомегаловирусом при переливаниях крови, донорским инфицированным молоком.
Из протозойных инвазий определенное значение в возникновении пороков имеет лишь токсоплазмоз. Пораженный при этом эмбрион обычно погибает, а у плода могут развиться вторичная микро- или гидроцефалия, микрофтальмия. Для каждого инфекционного заболевания не существует специфического и просто распознаваемого дефекта, однако при множественных пороках развития необходимо заподозрить внутриутробную инфекцию. Ее следует заподозрить у любого больного ребенка с небольшими размерами тела, не соответствующими гестацион-ному возрасту, т. е. с отставанием развития и микро- или гидроцефалией, нарушением зрения, катарактой и/или глаукомой, увеличенными размерами печени и селезенки. Однако внутриутробные инфекции отличаются широким спектром клинических проявлений: новорожденный может страдать множественными пороками развития.
Формирование пороков происходит преимущественно в период эмбрионального морфогенеза (3-10-я неделя беременности) в результате нарушения процессов размножения, миграции, дифференциации и гибели клеток. Эти процессы происходят на внутриклеточном, экстраклеточном, тканевом, межтканевом, органном и межорганном уровнях. Нарушением размножения клеток объясняют гипоплазию и аплазию органов. Нарушение их миграции лежит в основе гетеротопий. Задержка дифференциации клеток обусловливает незрелость или персистирование эмбриональных структур, а ее полная остановка – аплазию органа или его части. Нарушение физиологической гибели клеток, как и нарушение механизмов адгезии (“склеивание” и срастание эмбриональных структур), лежат в основе многих дизрафий (например, спинномозговых грыж).
Выделяют несколько групп пороков. В зависимости от времени воздействия вредных факторов и объекта поражения выделяют следующие формы пороков развития.
1. Гаметопатии – патологические изменения в половых клетках, произошедшие до оплодотворения и приводящие к спонтанному прерыванию беременности, врожденным порокам развития, наследственным заболеваниям. Это наследственно обусловленные врожденные пороки, в основе которых лежат спорадические мутации в половых клетках родителей или унаследованные мутации у более отдаленных предков.
2. Бластопатии – это повреждения зиготы в первые 2 недели после оплодотворения (до момента завершения дифференциации зародышевых листков и начала маточно-плацентарного кровообращения), вызывающие гибель зародыша, внематочную беременность, пороки развития с нарушением формирования оси зародыша (симметричные, асимметричные и неполностью разделившиеся близнецы, циклопия, аплазия почек и др.).
3. Эмбриопатии – поражения зародыша от момента прикрепления его к стенке матки (15-й день после оплодотворения) до сформирования плаценты (75-й день внутриутробной жизни), проявляющиеся пороками развития отдельных органов и систем, прерыванием беременности. Поскольку в эмбриональный период происходит формирование основных морфологических структур органов, то естественно, что большинство врожденных пороков образуется именно в этот период. Наличие критических периодов, т. е. стадий интенсивной дифференцировки органов, когда они наиболее легко повреждаются, определяет существование временной специфичности для различных органов. Так, воздействие повреждающего фактора на 4-6-й неделе внутриутробного развития часто ведет к формированию у плода порока сердца, на 12-14-й неделе – порока развития половых органов и т. д. Локализация дефекта также зависит от интенсивности повреждающего воздействия.
4. Фетопатии – общее название болезней плода, возникающих под воздействием неблагоприятных факторов с 11-й недели внутриутробной жизни до начала родов. Важнейшая роль в формировании фетопатии принадлежит состоянию плацентарного комплекса. Признаками фетопатии становятся: задержка внутри- утробного развития; врожденные пороки в результате обратного развития зародышевых структур (кишечный свищ, открытые артериальный проток или овальное окно) или эмбриональных щелей (расщелины губы, неба, позвоночника, уретры); сохранение первоначального расположения органов (крипторхизм); гипоплазии и дисплазии отдельных органов и тканей (дисплазия почек, микроцефалия, гидроцефалия и др.); избыточное разрастание соединительной и других тканей при инфекциях (катаракта и др.); врожденные болезни (гемолитическая болезнь новорожденных, гепатиты, циррозы, пневмонии, миокардиты, энцефалиты и др.). Фетопатии нередко приводят к преждевременным родам, асфиксии при рождении, метаболическим и другим нарушениям адаптации новорожденных к внеутробной жизни и являются наиболее частыми причинами неонатальных болезней и смертности.
К врожденным порокам относятся следующие нарушения развития.
1. Агенезия – полное врожденное отсутствие органа.
2. Аплазия – врожденное отсутствие органа или выраженное его недоразвитие. Отсутствие некоторых частей органа называется термином, включающим в себя греч. слово olygos (“малый”) и название пораженного органа. Например, олигодактилия – отсутствие одного или нескольких пальцев.
3. Гипоплазия – недоразвитие органа, проявляющееся дефицитом относительной массы или размеров органа.
4. Гипотрофия – уменьшенная масса тела новорожденного или плода.
5. Гиперплазия (гипертрофия) – повышенная относительная масса (или размеры) органа за счет увеличения количества (гиперплазия) или объема (гипертрофия) клеток.
6. Макросомия (гигантизм) – увеличенные длина и масса тела. Термины “макросомия” и “микросомия” нередко применяются для обозначения соответствующих изменений отдельных органов.
7. Гетеротопия – расположение клеток, тканей либо целых участков органа в другом органе или в тех зонах того же органа, где их быть не должно.
8. Гетероплазия – расстройство разграничения некоторых видов ткани. Гетероплазии следует дифференцировать от ме-таплазий – вторичного изменения разграничения тканей, которое связывают с хроническим воспалением.
9. Эктопия – смещение органа, т. е. локализация его в не- свойственном ему месте. Например, наличие почки в тазу, серд- ца – вне грудной клетки. Удвоение и увеличение в числе того или иного органа или части его.
10. Атрезия – полное отсутствие канала или естественного отверстия.
11. Стеноз – сужение канала или отверстия.
12. Неразделение (слияние) органов двух симметрично или асимметрично развитых однояйцевых близнецов. Название пороков, определяющих неразделение конечностей или их частей, начинается с греч. приставки syn (“вместе”) – синдактилия, симподия (соответственно – неразделение пальцев и нижних конечностей).
13. Персистирование – обратное развитие морфологических структур, которые в норме исчезают к определенному периоду развития (артериальный проток или овальное окно у ребенка в возрасте старше 3 месяцев). Одной из форм персистирования является дизрафия (арафия) – незаращение эмбриональной щели (расщелины губы, неба, позвоночника и т.д.).
14. Дисхрония – нарушение темпов (ускорение или замедление) развития.
Процесс может касаться клеток, тканей, органов или всего организма. Врожденные пороки могут проявляться и другими изменениями органов. Например, нарушением лобуляции (увеличение или уменьшение долей легкого или печени), образованием врожденных водянок (гидроцефалия, гидронефроз), инверсией – обратным (зеркальным) расположением органов. В зависимости от последовательности возникновения различают первичные и вторичные пороки. Первые непосредственно связаны с мутациями или воздействием тератогенных факторов. Вторые являются следствием первичных пороков (гидроцефалия, развившаяся при спинномозговой грыже) или обусловлены альтернативно-пролиферативными процессами в нормально развивающихся органах (гидроцефалия при токсоплазмозе). Выделение первичных пороков из комплекса обнаруженных у ребенка нарушений развития имеет большое значение для медико-генетического прогноза, поскольку риск определяется по основному пороку.
В связи с распространенностью пороки классифицируют на изолированные, системные и множественные. Изолированными называют первичные пороки, которые отмечаются лишь в каком-либо одном органе (микроцефалия, шестипалость). Системные пороки объединяют несколько первичных пороков в одной системе органов (ахондроплазия).
Множественные пороки составляют группу первичных пороков и дисплазии, отмечающиеся в двух и более системах органов (гидроцефалия в сочетании с дисплазиями лица и шестипалостью). Множественные пороки в свою очередь подразделяются на синдромы и неклассифицированные комплексы.
Под синдромами понимают устойчивые сочетания нескольких первичных пороков, например COFS-синдром (церебро-окуло-фацио-скелетный), основными признаками которого являются микроцефалия, микрофтальмия, катаракта, множественные дис-плазии лица, скелетные аномалии (вывихи в суставах, сгибатель-ные контрактуры) и ряд пороков других органов.
К неклассифицированным комплексам относят пороки, проявления которых не укладываются ни в один:
1) изменением наследственных структур (мутациями);
2) воздействием тератогенных факторов;
3) воздействием и мутаций, и тератогенных факторов (пороки мультифакториального генеза).
Среди пороков центральной нервной системы (ЦНС) различают пороки конечного мозга, обонятельного анализатора, стволовых отделов, мозжечка, спинного мозга и позвоночника, вентрикулярной системы и субарахноидального пространства. неустановленной этиологии.